Activation-induced cell death (AICD) is part of the immune system’s important checks and balances, but as with multiple other regulatory pathways, tumors can co-opt T cell AICD as a mechanism of immune escape. To better understand how tumors induce AICD and which immune cells are targeted, Huang et al. performed an in-depth discovery and mechanistic analysis and published their results in Nature Immunology.
To begin, Huang et al. evaluated immune cell subsets from breast and lung cancer patients and evaluated their sensitivity to AICD. While tumor infiltrating Tregs and type 2 T helper (TH2) cells were found to be fairly resistant to tumor-induced apoptosis, effector cytotoxic T lymphocytes (CTLs) and type 1 T helper (TH1) cells underwent apoptosis in higher proportions. Confirming this observation in vitro using isolated TILs, CD3 stimulation or exposure to tumor cell antigens induced more apoptosis in tumor-specific CTLs and TH1 cells than in Tregs or TH2 cells. Interestingly, the expression of death receptors including Fas, TNFR2, and TRAILR, was comparable between the different T cell subsets, indicating that despite an abundance of the cognate ligands (FasL, TNF, TRAIL) in these tumors, internal signaling was likely responsible for the differential sensitivity to AICD.
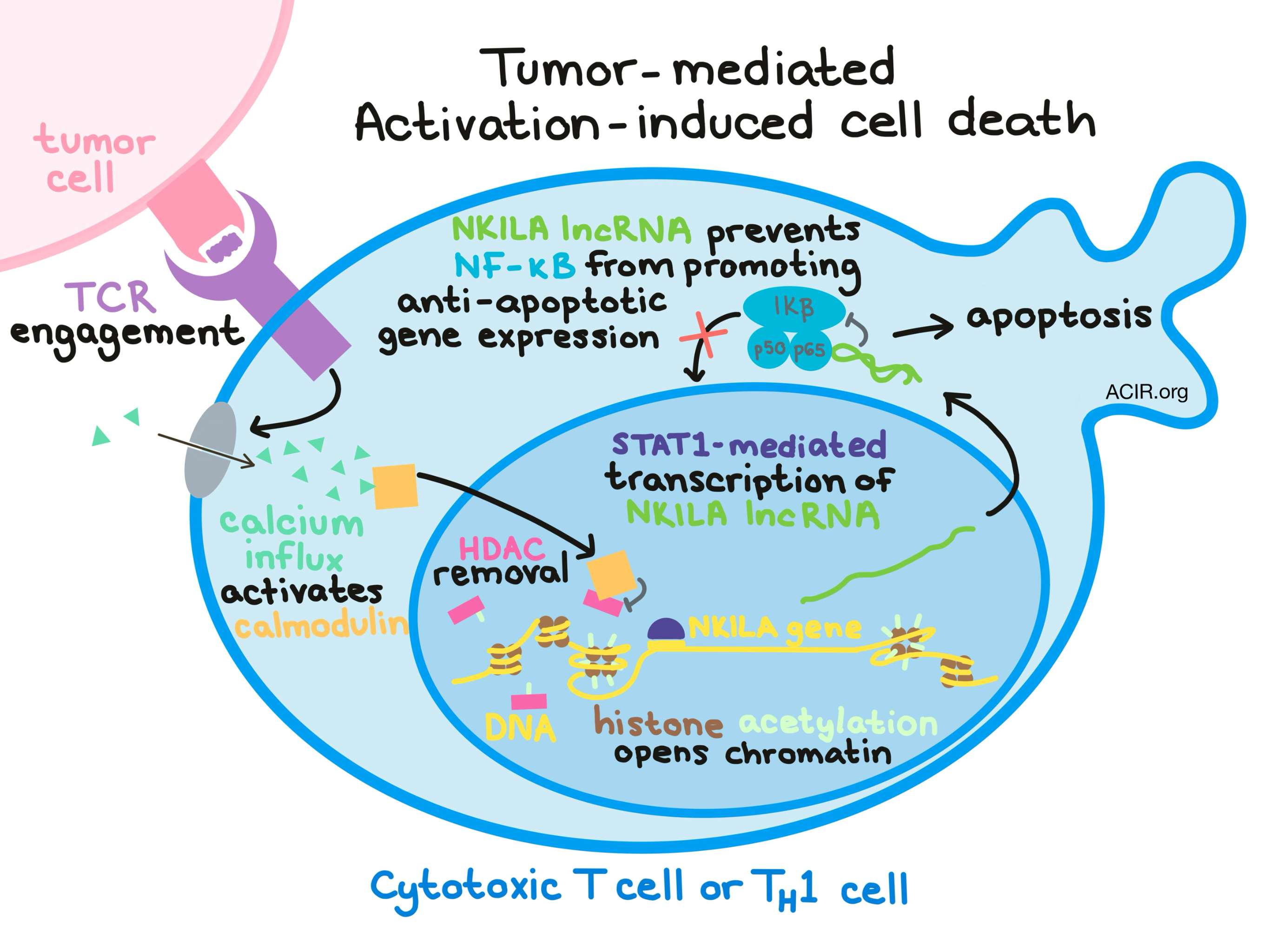
Diving into the mechanism underlying sensitivity to AICD, Huang et al. used gene enrichment analysis and uncovered that rather than being death receptor-dependent, AICD sensitivity was instead dependent on the activity of the NF-κB – a transcription factor that is essential for cytokine production and cell survival. Next, hypothesizing a role for long non-coding RNA (lncRNA), they identified the NKILA lncRNA as an inhibitor of NF-κB activity that functions by binding directly to the p65 protein within the NF-κB-IκBα complex. NKILA lncRNA was found to gradually increase during CTL activation and was most upregulated during the late AICD-sensitive phase. Further, upregulated NKILA was observed in CTLs and TH1 cells, but not in Tregs or TH2 cells, from patient-derived breast and lung cancer samples. In vitro silencing of NKILA with short hairpin RNA (shRNA) in stimulated T cells decreased apoptosis in activated cells, and forcing NKILA overexpression in Tregs or TH2 cells enhanced cells’ sensitivity to tumor-mediated AICD. The NKILA lncRNA-induced sensitivity to AICD was dependent on its ability to inhibit NF-κB.
Once NKILA and NF-κB were confirmed as key players in the mechanism of tumor-mediated AICD, the researchers began to piece together the early events in the tumor-induced mechanism of NKILA upregulation. They found that TCR signaling was critical to induction of AICD and confirmed that TCR signaling leads to an influx of calcium, which results in nuclear translocation of calmodulin – a calcium-modulated protein that mediates multiple cellular events. Next, they demonstrated that calmodulin binds to and inhibits histone deacetylases (HDACs), causing the HDACs to be removed from the NKILA promoter. Upon removal of the HDACs, histone acetyl transferases were recruited to acetylate the histones of the NKILA promoter, thus opening up the chromatin and making it available for transcription. With the chromatin open, STAT1 was able to move in and bind to the promoter at an identified STAT1 binding site, allowing for STAT1-mediated transcription of NKILA. As previously shown, the NKILA lcnRNA then interacts with NF-κB and inhibits its activity, rendering T cells incapable of performing essential cell survival functions and ultimately causing cell death.
To demonstrate the potential clinical relevance of NKILA-induced AICD in cancer, the researchers observed that in patients with breast cancer, high NKILA expression in tumor-specific CTLs from peripheral blood correlated with less infiltration of those tumor-specific CTLs into tumors. Further, patients with a high percentage of NKILAhi tumor-specific CTLs had shorter disease-free and overall survival.
Examining a possible avenue for therapeutic intervention, the Huang et al. transduced CD8+ T cells with NKILA shRNA to silence NKILA expression. When these cells were adoptively transferred into mice bearing patient-derived breast cancer xenografts, the transferred CTLs trafficked to the tumor and underwent less apoptosis, resulting in dramatic tumor cell apoptosis and inhibition of tumor growth compared to mice treated with adoptive transfer of control cells. Further, NKILA silencing upregulated CD107a and perforin, indicating enhanced cytotoxicity of the adoptively transferred cells, and increased the percentages of CD8+CD45RO+ effector T cells infiltrating the tumors.
Overall, Huang et al. identified the T cell subsets that are most susceptible to AICD and presented a thorough understanding of the underlying mechanism that controls this enhanced sensitivity. Further, the researchers presented evidence that this pathway is relevant in the clinical setting and that this mechanism may have the potential to be exploited for immunotherapy.
by Lauren Hitchings



