Human cytomegalovirus (HCMV) infection can have long-term immune effects, and NK and T cells both play roles in keeping HCMV latent. Based on the knowledge that CD8+ T cells can display NK cell-like properties, and that these cells can have a beneficial presence in individuals infected with particular intracellular pathogens causing tuberculosis or leprosy, Sottile et al. investigated this subset and determined the phenotype, transcriptomics, and reactivity of NKG2C+CD8+ T cells after HCMV exposure. Their results were recently published in Science Immunology.
The researchers started by assessing peripheral blood from 331 healthy donors to quantify NKG2C+CD8+ T cells. Out of these, 64% were HCMV seropositive, and in some individuals, NKG2C was expressed on T cells and NK cells. Of the seropositive donors, 41.5% had an expanded population of NKG2C+CD8+ T cells, with frequencies between 2.6% and 27.8%. While all donors had some NKG2C+CD8+ T cells present, they accounted for less than 2% of CD8+ T cells in seronegative donors. In those with expanded NKG2C+CD8+ T cells, NKG2C was not expressed on HCMV-reactive CD8+ T cells reacting to the dominant pp65 HCMV antigen, and the population expressing NKG2C+ did not show reactivity against the most commonly recognized HCMV epitopes.
The researchers then moved to a cohort of patients who had undergone umbilical cord hematopoietic cell transplants, as this is a clinical setting with a high incidence of HCMV reactivation. In temporal samples of this cohort, there was an expansion of the NKG2C+CD8+ T cell population after HCMV reactivation – an effect not seen in patients not experiencing reactivation.
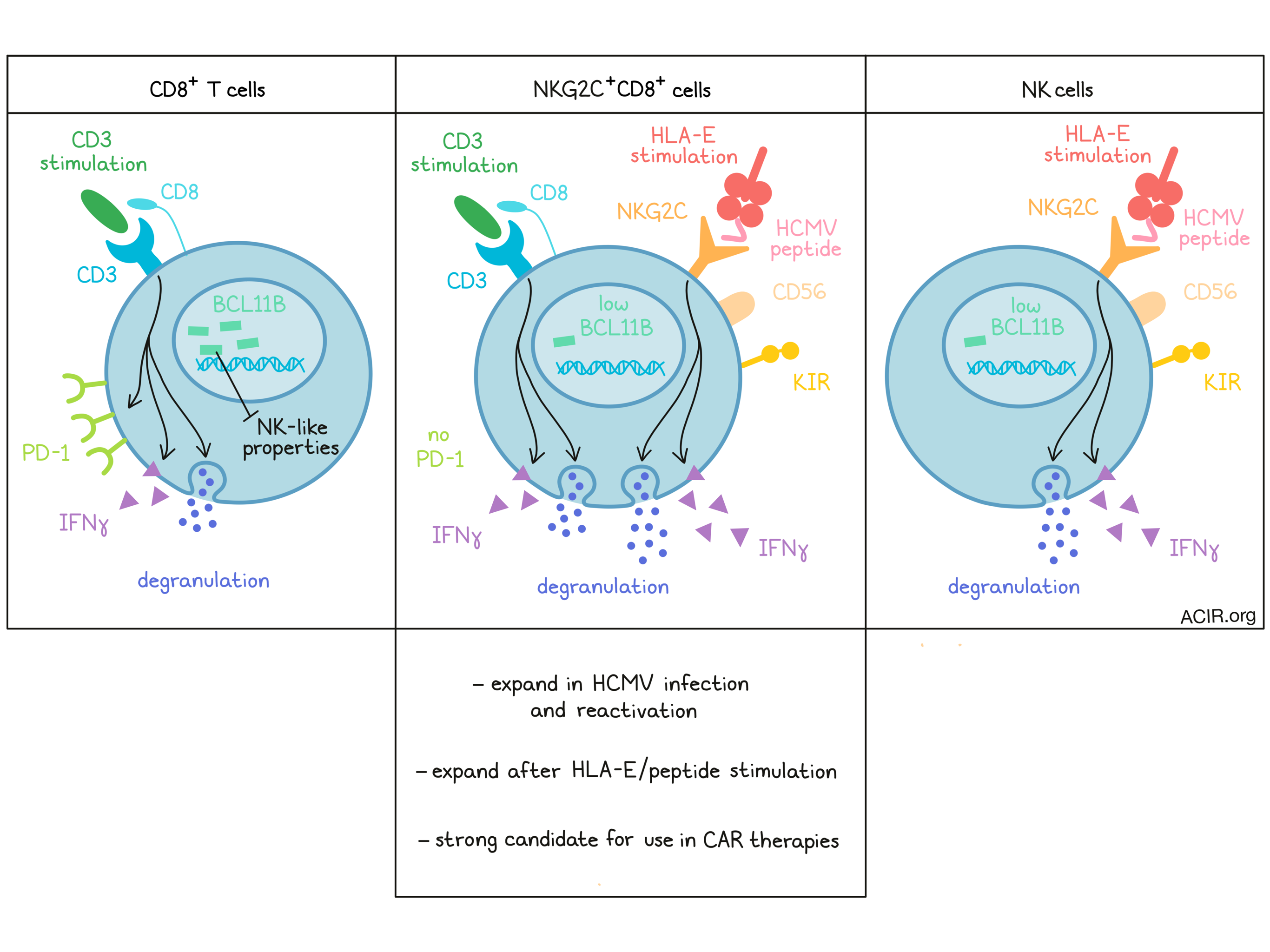
Further phenotyping of the NKG2C+CD8+ T cell population revealed that these cells coexpressed CD56 and inhibitory killer immunoglobulin-like receptors (KIRs) – canonical NK cell markers. The population also presented a phenotype resembling terminally differentiated T effector memory RA (Temra) cells. Overlap of the T cell differentiation expression patterns in the total NK population and in NKG2C+CD8+ T cells suggested the NKG2C+CD8+ T cell population might be NK-like.
Assessing whether NKG2C+CD8+ T cells, like Temra, expressed markers associated with terminal differentiation, the researchers discovered that this population did not express PD-1. After in vitro stimulation with CD3/CD2/CD28, the cells showed similar proliferation kinetics as similarly stimulated NKG2C-CD8+ T cells, but remained negative for PD-1.
To assess the transcriptional differences between the CD8+ T cell populations positive and negative for NKG2C, whole transcriptome analysis was conducted on paired peripheral blood samples from healthy HCMV-seropositive donors. The NKG2C+ population was enriched for genes associated with the NK cell-mediated cytotoxicity pathway, and expressed higher levels of NK cell-associated genes and lower levels of T cell-associated genes. Among the most differentially expressed genes, the transcription factor BCL11B stood out. This gene is expressed in all T cell subsets and progenitors, regulates key processes in T cell function and survival, and prevents the development of lymphocytes toward an NK-like phenotype. The NKG2C+ cells expressed lower levels of BCL11B transcripts, suggesting a loss of T cell identity. Additionally, the human orthologs of the Bcl11b-dependent genes found in mice that were knocked out for Bcl11b were also found in the human NKG2C+CD8+ T cells. NK cells expressed similarly low levels of BCL11B as the NKG2C+CD8+ T cells.
Comparing gene signatures, NKG2C+CD8+ T cells showed high expression of NK cell signatures, but no clear Temra signature expression. This suggested that NK cells and NKG2C+CD8+ T cells were closely related both at the phenotypic and the transcriptional level.
Next, NKG2C-CD8+ T cells from HCMV- humans were knocked out for BCL11B and expanded using either CD3/CD2/CD28 or K562 cells expressing the NKG2C target (HLA-E with a high-affinity HCMV peptide (HLA-E:VMAPRTLFL)). Among the knockout cells, more cells expressed CD56, but not PD-1, and there was a preferential expansion of CD45RA+CCR7- cells. After stimulation with the K562 cells, NKG2C+ cells reactive against the K562 cells emerged when BCL11B was knocked out – an effect not seen among cells stimulated with CD3/CD2/CD28.
Next, Sottile et al. found that in vitro stimulation of NKG2C with HLA-E stimulated degranulation and IFNγ production in NKG2C+CD8+ T cells. The same cells also responded to CD3 stimulation, like NKG2C-CD8+ T cells. When both NKG2C and CD3 were stimulated, there was a further increase in IFNγ production, suggesting co-triggering was possible and might induce higher responsiveness.
NKG2C+CD8+ T cells were reactive against HCMV-infected fibroblasts, but unreactive to uninfected cells. Also, when co-incubated with the K562 HLA-E:VMAPRTLFL cells, there was reactivity, measured by CD107a mobilization (indicative of degranulation) and IFNγ production, resulting in the killing of the cells. Blockade of NKG2C did not completely limit the effects, suggesting TCR signaling might also play a role. Therefore, the researchers investigated TCR Vβ usage of these cells in six people. In all samples, most of the NKG2C+ population expressed one prominent TCR Vβ chain, while there was a diversity in other populations. In half of the samples, there was a skewed distribution toward the Vβ chain TRBV-14. Only the NKG2C+CD8+ T cells with TRBV-14 usage had a higher killing capacity against leukemia cell lines than NKG2C-CD8+ T cells. This could be partially inhibited by CD94 blockade (CD94 is a co-receptor of NKG2C). The involvement of the TCR was investigated by knocking out TCRs, which did not result in a reduction in CD107a mobilization and IFNγ production. However, loss of HLA-E expression or blockade of HLA-E binding abrogated the reactivity, and tetramer binding suggested that the TCRs recognized HLA-E presenting non-self peptides.
Co-incubation of PBMCs with K562 HLA-E:VMAPRTLFL cells increased the frequency of NKG2C+CD8+ T cells, and NKG2C+CD8+ T cells could expand rapidly when the target cells expressed HLA-E. When these expanded cells were transduced with a CD19 CAR construct, co-culturing with the CD19+ cell line NALM-6 revealed a higher killing capacity than the CAR T cells derived from NKG2C-CD8+ T cells. In addition, the NKG2C+ cells did not upregulate PD-1, and even cells not transduced with the CAR could recognize the NALM-6 cells, suggesting additional and complementary reactivity when NKG2C+CD8+ T cells were used for CAR T cell therapy.
These data suggest that this NK-like T cell population might induce a greater cytotoxicity potential, a mechanism that has potential to be exploited to improve the efficacy of cellular immunotherapies.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Rosa Sottile from The Katharine Hsu Lab at Memorial Sloan Kettering Cancer Center answered our questions.

What prompted you to tackle this research question?
Our team, led by physician-scientist Katharine Hsu, was always interested in how a virus called human cytomegalovirus (CMV) can shape the natural killer cell (NK)-mediated immune response to viral infected cells and tumors. By studying healthy volunteers and transplant patients, we noticed that a population of unusual T cells emerge in the blood after CMV infection. These unusual T cells share many features with NK cells, challenging the concept of a clear distinction between innate and adaptive immunity. We found this virus-mediated “identity shift” very fascinating and continued to explore the properties of these hybrid cells to gain more insights into immune cell plasticity and how to take advantage of it in the battle against cancer.
What was the most surprising finding of this study for you?
This T cell population has lost a protein, called BCL11B, believed to be essential for normal T cell development. We found that late during their life cycle, some already mature T cells can divert naturally towards a more innate-like identity by losing BCL11B. This means that these cells can also acquire functioning NK receptors and recognize targets with a “double sword” mechanism. By modulating BCL11B, we could potentially generate this hybrid cell de novo from any donor and use it as a starting point to create a new immunotherapy treatment. The current T cell-based immunotherapies have some disadvantages, including vulnerability to be shut down by immune checkpoints like PD-1. Our hybrid cell is resistant to PD-1 upregulation, thus making it an attractive candidate for adoptive cell therapies, like CAR-T.
What was the coolest thing you’ve learned (about) recently outside of work?
You can sneeze 5 times faster than Usain Bolt can run!



