Mainly due to its location in the pons, a part of the brainstem that controls many of the body's vital functions, and its infiltrative nature, diffuse intrinsic pontine glioma (DIPG) is one of the most devastating and challenging childhood cancers. Currently, oncologists can only offer radiotherapy to stabilize the tumor and reduce symptoms. Sadly, over 90% of the young patients die within two years of diagnosis. In a recent paper published in Cancer Cell, Ausejo-Mauleon et al. suggest TIM-3 blockade as a possible effective treatment option for DIPG, and push for future clinical evaluation.
First, to confirm the potential for therapeutic targeting of TIM-3, Ausejo-Mauleon and team performed in silico analysis of DIPG patient samples, validating TIM-3 expression in 94% of cases and no expression in healthy human brains. Relative expression levels of TIM-3 were higher than those of other immune checkpoint molecules, such as CTLA-4, PD-1, LAG-3, and TIGIT, and expression of the main TIM-3 ligand, galectin-9, was also increased. Single-cell analysis revealed TIM-3 expression in all three different DIPG tumor cell populations: astrocyte-like, oligodendrocyte-like, and oligodendrocyte precursor cell (OPC)-like. OPC-like cells showed the highest expression of TIM-3, and gene enrichment analysis suggested a direct role of TIM-3 in tumor cell survival and tumorigenesis.
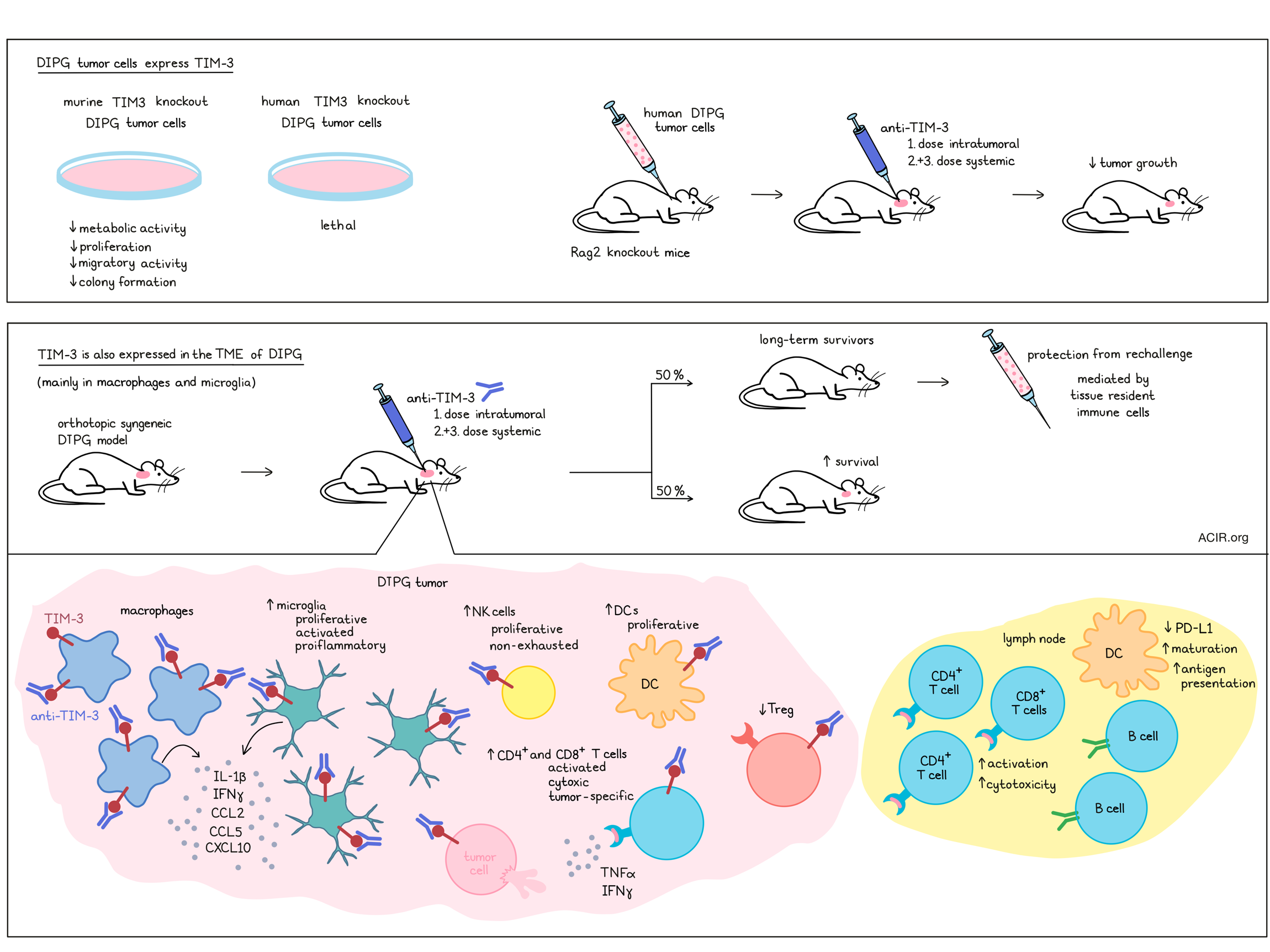
Three murine DIPG cells lines with TIM-3 knockout (KO) were viable, but showed decreased metabolic activity, proliferation, migratory activity, and capability to form colonies. Immunocompetent mice orthotopically implanted with TIM-3 KO cell lines in the pons of the brainstem had a longer median survival than mice implanted with parental cells. This effect was due to a slower growth rate of TIM-3 KO DIPG cells, and not due to differences in immune infiltrates. Transcriptomic analysis revealed downregulation and/or decreased phosphorylation of components of the MAPK and PI3K-AKT signaling pathways in TIM-3 KO DIPG cells compared to parental cells.
In human DIPG cell lines, genetic ablation of TIM-3 was lethal, and knockdown of TIM-3 expression with doxycycline-inducible shRNA or TIM-3 blockade with anti-TIM-3 (BMS-986258) reduced viability of the evaluated human DIPG cell lines. In vivo, immunodeficient Rag2 KO mice orthotopically implanted with a human DIPG cell line and treated with anti-TIM-3 showedda longer median survival of 78 days, compared to 63 days for mice treated with a control antibody.
In addition to the DIPG tumor cells themselves, TIM-3 is also expressed, at even higher levels, in the immune compartment of DIPG, as revealed by in silico analysis of scRNAseq data for DIPG patients. The tumor microenvironment (TME) of DIPG is mainly composed of microglia and macrophages, with nearly negligible numbers of T cells. Multiplex immunofluorescence analysis of 9 patient samples demonstrated TIM-3 expression in 54% of CD68+ myeloid cells, 39% of CD3+ T cells, and 44% of H3K27M+ DIPG tumor cells. Of note, TIM-3 displayed a differential expression pattern depending on H3 status (higher in mutated H3). In microglia, TIM-3 expression correlated with genes involved in the activation of an immune response. When the TME of DIPG in an immunocompetent model was assessed 10 days after implantation, TIM-3 expression was found on 85% of CD8+ T cells, 79% of DCs, 69% of microglia, 66% of macrophages, 63% of Tregs, 61% of CD4+ T cells, 55% of monocytes, and less than 50% of NK cells and granulocytes. The highest expression intensity was observed for DCs, macrophages, microglia, and Treg populations.
Ausejo-Mauleon and colleagues treated immunocompetent DIPG mice with anti-TIM-3 on a hybrid schedule, in which an initial intratumoral administration three days after tumor implantation was followed by two systemic doses. Biodistribution analysis by SPECT scan revealed antibody in the brainstem, which declined over the course of one week, and antibody in the tumor-draining cervical lymph nodes, which are located in the neck region. Treatment led to an increase in median overall survival, with 50% long-term survivors, which were also able to reject a new tumor implantation. When this tumor rechallenge experiment was performed in the presence of FTY720, abrogating T cell traffic from the lymph nodes, survivors were still able to reject the repeated tumor challenge, suggesting that resident memory cells were responsible for tumor rejection. Of note, systemic-only administration also resulted in a significant, but less pronounced, therapeutic benefit. Treatment was well tolerated, and weight loss or hepatic toxicity were not observed. Seven days after the first treatment dose, the tumor site was characterized by a 3-fold increase in microglia with a proliferative phenotype and by increases in the percentage of proliferative, non-exhausted NK cells and proliferative DCs. Intratumoral levels of IL-1β and IFNγ, and chemoattractants CCL2, CCL5, and CXCL10 were increased, as were the number of infiltrating tumor-specific CD8+ T cells. CD8+ and CD4+ T cells displayed an activated, cytotoxic IFNγ+TNFα+ or GrzB+TNFα+ phenotype upon restimulation. By day 14, augmentation of an adaptive immune response was observed, with increases in the ratios of proinflammatory CD8+ T cell:Treg, CD4+ T cell:Treg and CD8+ T cell:macrophage.
Concurrent depletion of NK, CD4+, and CD8+ T cells resulted in a partial loss of the therapeutic effect of anti-TIM-3 treatment, and only administration of the CSF1R inhibitor, PLX-3397, led to a total loss of survival benefit. PLX-3397 preferentially depleted macrophages and decreased the number of active, proliferating Ki67+MHC-II+ microglia, and CD4+ and CD8+ T cells. At the same time, the upregulated levels of IL-1β, IFNγ, CCL2, CCL5, and CXCL10 were lost. Thus, TIM-3 blockade resulted in the activation of proinflammatory macrophages and microglia, which induced pro-inflammatory cytokines and chemokines that in turn enhanced antitumor T cell responses.
Next, the researchers set out to study the kinetics of different immune populations in the deep cervical and superficial cervical tumor draining lymph nodes (LN). At day 7 after the first treatment dose, deep cervical LNs of anti-TIM-3-treated mice had significantly higher numbers of CD45+ immune cells due to increases in the CD4+ and B cell populations, and to a lesser extent, in CD8+ T cells and DCs. DCs in the deep cervical LN displayed a significantly lower expression of PD-L1, and DCs in the superficial cervical LN had a mature, antigen-presenting CD80+MHCII+ phenotype. In particular, the deep, but also the superficial cervical LN demonstrated significantly higher percentages of activated, cytotoxic CD4+ and CD8+ T cells with a Ki67+TNFα+ and GrzB+TNFα+ phenotype upon anti-TIM-3 treatment. The researchers assumed that effective antigen presentation by DCs mediated T cell activation and proliferation.
Interestingly, the value of targeting TIM-3 appeared to be unique to DIPG. Despite expression of TIM-3 in GBM patients, valuation of anti-TIM-3 in one orthotopic TIM-3+ glioblastoma model (CT-2A) led to no therapeutic benefit. And although PD-1 expression was also confirmed in the orthotopic DIPG model used here, anti-PD-1 treatment was not effective.
Here, Ausejo-Mauleon and colleagues showed that TIM-3 blockade can have a direct effect on DIPG tumor cells. Furthermore, in immunocompetent, orthotopic DIPG models, anti-TIM-3 transformed the TME toward a more proinflammatory milieu and promoted a coordinated action of mainly macrophages, microglia, and CD8+ T cells to eliminate tumor cells and produce a durable memory response. This study establishes TIM-3 blockade as a new therapeutic approach for DIPG, and urges for the clinical evaluation of this new glimmer of hope.
Write-up by Ute Burkhardt, image by Lauren Hitchings and Ute Burkhardt
Meet the researcher
This week, first author Iker Ausejo-Mauleon answered our questions.

What was the most surprising finding of this study for you?
What surprised us most was the high expression of a typical T cell immune checkpoint molecule such as TIM-3 in these pediatric brain tumors in comparison with other molecules, such as PD-1, CTLA-4, LAG-3, or TIGIT. Although we already knew about TIM-3 expression in myeloid cells, we were surprised by its high expression in tumor-infiltrating macrophages and microglia. Moreover, it was fascinating to find out that this high expression in many immune cell types meant that blocking it increased survival and led to long-term survivors and immune memory in orthotopic models of DIPG, where we had not previously seen these good results with other therapies.
What is the outlook?
TIM-3 blockade emerges as an exciting alternative to classical immune checkpoints, such as PD-1, that did not obtain the desired results in clinical trials for DIPG and other pediatric brain tumors. Moreover, the lack of other effective therapies for these devastating pediatric brain cancers makes these preclinical results especially promising in the field, and offers a strong rationale for initiating a clinical trial with an anti-TIM-3 antibody for the treatment of DIPG.
What was the coolest thing you’ve learned (about) recently outside of work?
IAM: In the last months, I have learned how important it is to take care of myself mentally and to disconnect from my science life as well. For this, I like to go running, biking, climbing, and skiing in the mountains near my village (Arroniz, Spain) and in the Pyrenees. Discovering new paths, adventures, and new challenges every day helps me to disconnect from the pressure that science research puts on us.



