Metabolic changes in T cells are often accompanied by changes in gene expression, functionality, and differentiation state. To better understand the connections between some of these changes, Raychaudhuri and Singh et al. recently explored how metabolic pathways that increase lactate production can lead to changes in lactylation – a histone modification of lysine residues – and uncovered how that contributed to the regulation of various changes in T cells following TCR activation.
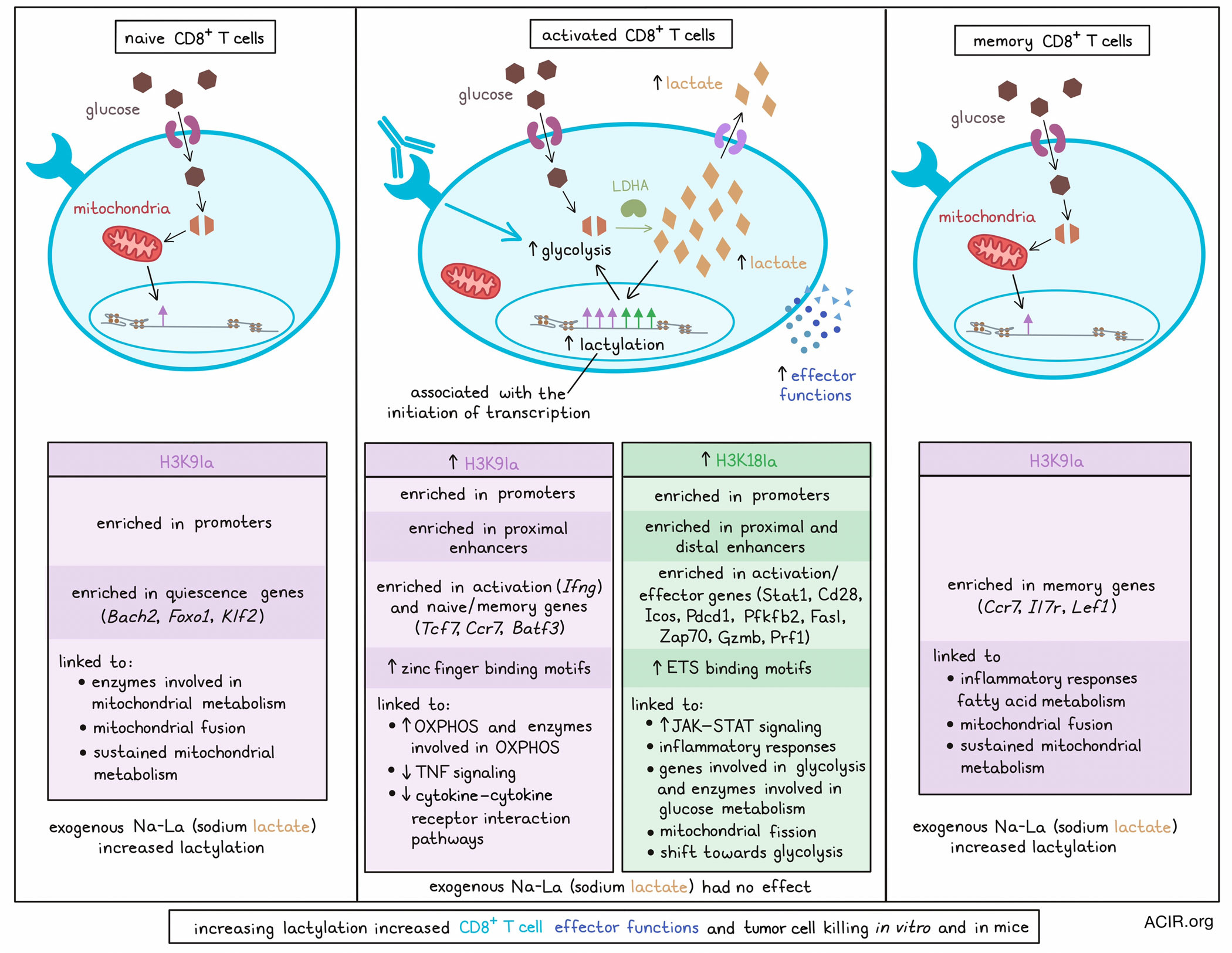
To begin, Raychaudhuri and Singh et al. evaluated histone lactylation patterns in naive and effector CD8+ T cells, and found that following activation with anti-CD3 or a cognate antigen, intracellular and extracellular lactate accumulated, as did H3K18 lactylation (H3K18la) and H3K9 lactylation (H3K9la), particularly in cells expressing effector markers like granzyme B, TBET, and IFNγ. This pattern of increased lactylation in highly activated cells was evident in cultures of both human and murine T cells, a murine GvHD model, a murine tumor model, and a murine tumor model treated with immune checkpoint blockade (anti-PD-1 + anti-CTLA-4).
To better understand the role of H3K18la and H3K9la in transcriptional regulation, the researchers performed ChIPseq and ChromHMM analysis to classify genomic regions into various states based on patterns of histone post-translational modifications (hPTMs), including modifications that initiated transcription, marked active enhancer regions, associated with transcribing gene bodies, and repressed transcription. This analysis showed that H3K18la and H3K9la were enriched near transcription start sites (TSSs) and CpG islands, and were associated with other transcription-initiating hPTMs in promoter regions and in proximal and distal (but not intergenic) enhancer-like sequence-bearing regions, suggested that lactylation was involved in initiating transcription.
In naive T cells, H3K9la was enriched in promoter regions and was increased following T cell activation. H3K18la, on the other hand, was minimally enriched in naive T cells, but was enriched on promoter regions following activation. Both H3K18la and H3K9la were enriched in proximal enhancer-like regions following T cell activation, but only H3K18la was enriched in distal enhancer-like regions. Regions marked by lactylation peaks also showed higher occupancy of RNApol II, suggesting enhanced transcription.
Integrating RNAseq data with their CHIPseq data, the researchers found that in activated CD8+ T cells, H3K18la (alone or in combination with other hPTMs) marked genes that were crucial for T cell activation and effector molecules (Stat1, Cd28, Icos, Pdcd1, Pfkfb2, Fasl, Zap70, Gzmb, and Prf1), while H3K9la was enriched in the T cell activation gene Ifng and in genes associated with naive and memory T cells (Tcf7, Ccr7, and Batf3). Transcription factor (TF) binding motif enrichment analysis further showed that H3K18la-marked promoter regions were highly enriched in ETS binding motifs, while H3K9la-marked promoter regions were enriched for zinc finger binding motifs; both ETS and zinc finger TFs are associated with CD8+ T cell differentiation and functionality. In naive CD8+ T cells, H3K9la was enriched in genes encoding molecules involved in T cell quiescence (Bach2, Foxo1, and Klf2), and in memory T cells, H3K9la was enriched in genes associated with memory (Lef1, Ccr7, and Il7r). Both H3K18la and H3K9la were limited in exhausted CD8+ T cells, and did not show enrichment in exhaustion-associated genes.
Next, the researchers used gene set enrichment analysis (GSEA) and found that in activated CD8+ T cells, H3K18la was linked to increased JAK–STAT signaling, inflammatory responses, genes involved in glycolysis, and enzymes involved in glucose metabolism, while H3K9la was linked to increased OXPHOS, enzymes involved in OXPHOS, decreased TNF signaling, and decreased cytokine–cytokine receptor interaction pathways. In memory T cells, H3K9la was linked to inflammatory responses and fatty acid metabolism, and in naive T cells, it was linked to enzymes involved in mitochondrial metabolism. H3K18la was also linked to mitochondrial fission during CD8+ T cell activation, whereas H3K9la was linked to mitochondrial fusion in naive and memory CD8+ T cells. Together, these findings suggest that H3K9la supports sustained mitochondrial metabolism, while H3K18la mediates the shift towards glycolysis during CD8+ T cell activation.
Compared to naive and memory T cell subsets, which rely mostly on OXPHOS and fatty acid oxidation, activated CD8+ T cells rely mostly on glycolysis, which leads to an accumulation of lactate. To determine whether this metabolic shift contributes to increased lactylation, the researchers used an LDHA inhibitor to inhibit lactate production, and found that this also inhibited H3K18la and H3K9la, and shifted mitochondrial morphology from fission towards fusion. Similarly, blocking lactate export increased lactylation in activated T cells, but not in memory T cells, where glycolysis and lactate accumulation were less prevalent. Instead, inhibition of the electron transport chain, fatty acid oxidation, or ATP citrate lyase reduced H3K9la in memory T cells, where these metabolic pathways were more dominant. Administration of exogenous sodium-L-lactate (Na-La) increased H3K18la and H3K9la in naive and memory, but not activated CD8+ T cells, dependent on lactate transport into the cells. In naive cells, Na-La increased H3K9la at gene promoters associated with naiveness, and drove increased expression of those genes.
To test whether modulation of H3K18la and H3K9la levels could be utilized to modulate the functionality of activated CD8+ T cells, the researchers treated them with LDHA, thereby inhibiting lactate production and subsequent lactylation. This reduced H3K18la and H3K9la on activation-associated gene promoters and reduced expression of those genes, ultimately reducing production of cytotoxic molecules and the capacity for antigen-specific T cell-mediated killing. Similar effects were observed with inhibition of CBP/EP300 – a known histone lactylase. However, targeting HDAC1–HDAC3 – known histone delactylases – with the HDAC inhibitor MS275 increased H3K18la and H3K9la, increasing the expression of effector genes and the capacity for antigen-specific T cell-mediated killing. In a mouse tumor model, treatment with MS275 increased H3K18la and H3K9la in tumor- and tdLN-derived CD8+ T cells, increased intratumoral granzyme B+ effector CD8+ T cells, and reduced tumor growth. Inhibition of HNO1, a histone lactylase, had the opposite effect.
This study begins to unravel the complex relationship between histone lactylation and metabolic, transcriptional, and functional changes that occur in T cells, particularly in response to TCR activation. Importantly, it also establishes modulation of histone lactylation as a potential strategy for enhancing the functionality of CD8+ T cells in tumor settings, opening up new avenues of exploration in cancer immunotherapy.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, lead author Sangeeta Goswami answered our questions.

What was the most surprising finding of this study for you?
One of the most surprising findings in our study was discovering how lactate, derived from different metabolic pathways, influenced specific histone modifications in various CD8+ T cell subsets. This raised an intriguing question: why do these metabolic sources selectively modify different histone residues? A major challenge we faced was that no specific writers and erasers of histone lactylation were known. So, we had to block multiple metabolic and epigenetic pathways to characterize the impact of histone lactylation in regulating T cell function. Additionally, our computational analysis showed that even though there were other histone modifications that regulated gene transcription, histone lactylation was critical for gene transcription in CD8+ T cells.
What is the outlook?
We expect our findings to shed light on how histone lactylation interacts with other histone modifications to regulate gene transcription in CD8+ T cells, which play an important role in fighting cancer and infection. Applying these insights to current clinical trials may also explain why some patients respond to immunotherapy while others don't, guiding new strategies to improve treatment outcomes.
What was the coolest thing you’ve learned (about) recently outside of work?
As a physician, scientist, mentor, and a mother, maintaining an optimum work-life balance is always an aspirational goal. However, playing each of these individual roles has taught me that they are interconnected, and that nothing brings greater joy than seeing the sense of happiness and pride in people I truly care about. For instance, seeing a therapeutic response in a patient, happiness in my mentees face on publishing a paper, or a sense of pride on my son’s face after his first orchestra concert.



