Patients whose tumors are strongly infiltrated by immune cells tend to have a better prognosis and better responses to immunotherapy, but the mechanisms that underlie immune infiltration are not clearly defined. To better understand the infiltration of T cells into tumors, Dangaj et al. investigated the roles of chemokines in the tumor microenvironment and identified an interesting sequence of events involving CCL5 and CXCL9. Their results were recently published in Cancer Cell.
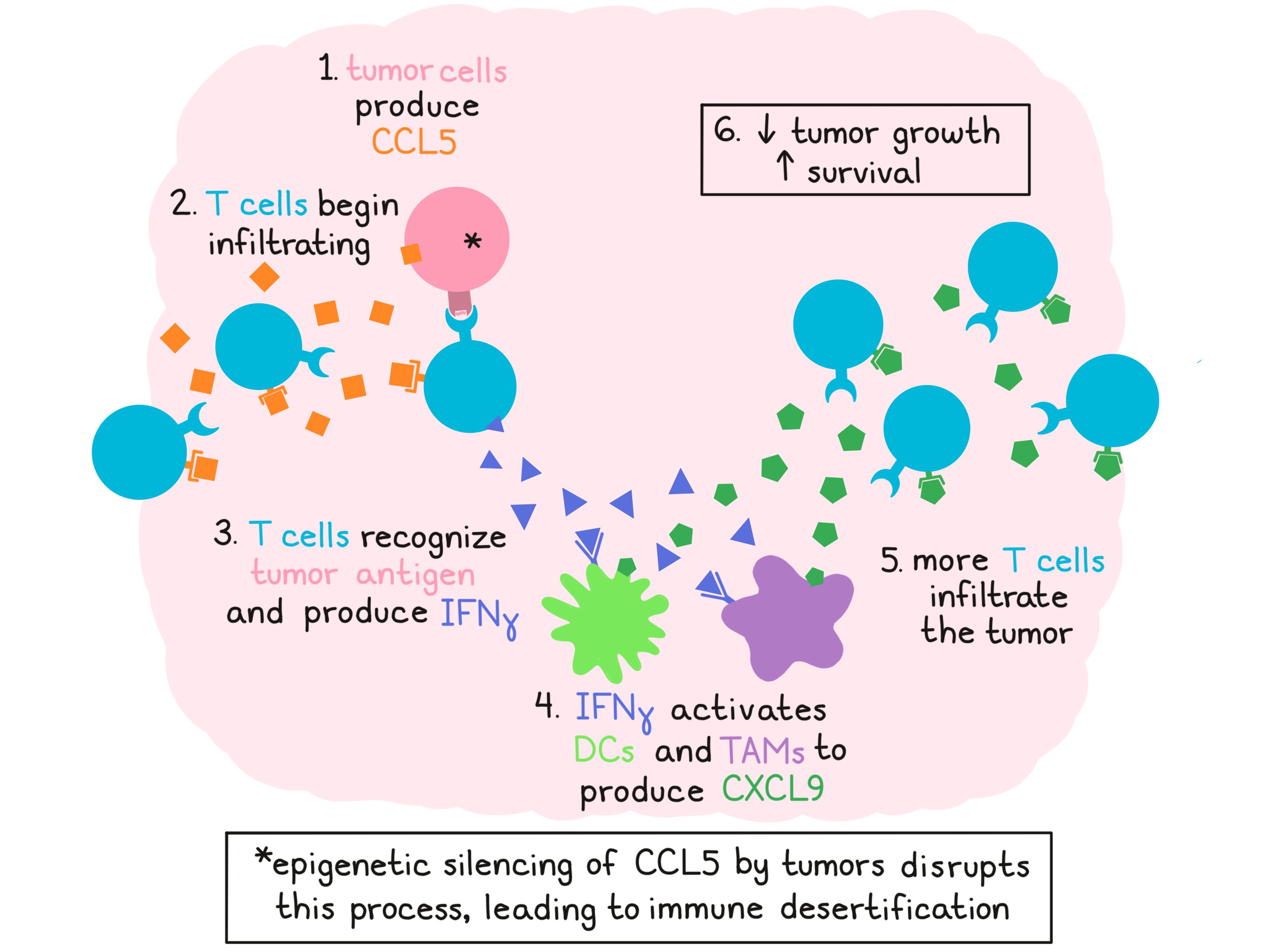
To begin, Dangaj et al. investigated chemokines associated with T cell infiltration in solid human cancers and found that CCL5 and CXCL9 uniquely correlated with T cell infiltration (as measured by CD8A expression) across several cancer types and multiple datasets. Using human epithelial ovarian cancer (EOC) as a model to investigate the role of these molecules in T cell engraftment, the researchers found that CCL5 was constitutively expressed by tumor cells and was associated with infiltration of intraepithelial (ie)CD8+ T cells. Further, CD8+ T cells enriched for the CCL5 receptor, CCR5, were identified in the proximity of CCL5+ tumor cells.
Although CCL5 was clearly related to T cell infiltration, not all CCL5+ tumors shared an immune-inflamed phenotype, suggesting that CCL5 was necessary, but not sufficient for T cell engraftment. Turning their attention toward CXCL9, the other chemokine which correlated with CD8A expression, the researchers found that CXCL9 was almost exclusively expressed in CCL5+ tumors, and that all tumors with high ieCD8+ T cell infiltration expressed CXCL9. Additionally, a large fraction of TILs in these tumors expressed the CXCL9 receptor, CXCR3. CXCL9 expression was localized around tumor islets (more so than within them) and in tertiary lymphoid aggregates in the stroma. CD68+ TAMs and CD11c+ DCs were identified as the primary sources of CXCL9. Expression of CXCL9 by these cell types correlated with IFNG and CD8A expression in the tumor. Using whole primary tumor cultures from EOC samples containing ieCD8+ TILs, the researchers showed that CXCL9 production by TAMs and DCs was dependent on IFNγ produced by TILs, and that the release of IFNγ by TILs was dependent on tumor antigen recognition. Based on these observations, CXCL9 could be considered a biomarker of T cell engraftment and activation in the TME.
Based on their observations, Dangaj et al. proposed that constitutive tumor expression of CCL5 initially attracts T cells to the tumor site, where they encounter tumor antigens and begin producing IFNγ. IFNγ then induces local TAMs and DCs to secrete CXCL9, which attracts another wave of infiltrating T cells. Dissecting this hypothesis, the researchers performed a chemotaxis assay on supernatants of primary TME cultures and showed that CCR5 blockade reduced TIL recruitment, and that the addition of CXCR3 blockade further reduced TIL recruitment. Using spheroids of OV79 ovarian cancer cells as an in vitro model of the EOC tumor microenvironment, researchers found that tumor infiltration was highest when tumors coexpressed antigen and CCL5, and the tumor microenvironment was enriched for IFNγ-activated CD11b+ monocytes.
Analysis of large datasets of EOC, triple-negative breast cancer, colon cancer, lung cancer, and melanoma showed that increased expression of CXCL9 was dependent on CCL5 expression, and that TIL infiltration was dependent on coexpression of CCL5 and CXCL9. Within each cancer type, patients whose tumors showed high expression of both CCL5 and CXCL9 survived longer and were predicted to have higher infiltration of CD8+ T cells, cytotoxic T cells, Th1 T cells, DCs, and macrophages. Gene pathway analysis revealed enrichment for TCR signaling, T cell activation, costimulation, and IFN signaling in these tumors. Overall, the researchers were able to identify a common 21-gene signature overexpressed in CCL5hiCXCL9hi tumors that was shared across multiple cancer types. In melanoma patients, overexpression of this gene signature in tumors prior to treatment was associated with response to treatment with nivolumab followed by ipilimumab, but not the reverse sequence (ipilimumab followed by nivolumab).
Looking into why some tumors are poorly infiltrated, the researchers found that tumor cells could epigenetically downregulate or silence CCL5 expression through DNA methylation, and that this was associated with reduced immune infiltration. In a murine syngeneic ID8 ovarian cancer model, which exhibits initial tumor infiltration with T cells followed by desertification within 6-7 weeks of tumor development, the researchers identified CCL5 silencing in late tumors, and the subsequent loss of CXCL9 and TIL engraftment. In line with this, early tumors were responsive to PD-1 blockade, while late tumors were resistant. Forced overexpression of CCL5 in this model resulted in increased Cxcl9, Cd8a, and Ifng, even in late tumors, resulting in reduced tumor growth and longer survival. Knockdown of CCL5, blockade of IFNγ, or blockade of CXCL9 had the opposite effect.
Overall, Dangaj et al. elucidate a mechanism by which CCL5 and CXCL9 coexpression governs immune infiltration. As CCL5 is often downregulated in tumors as an immune escape mechanism, reinvigoration of CCL5 expression could be a promising target for immunotherapy.
by Lauren Hitchings
Meet the Researcher
This week, we interviewed first author Denarda Dangaj and lead author George Coukos.
What prompted you to tackle this research question?
Since our initial discovery (Zhang et al., 2003) that ovarian tumors are infiltrated by CD8+ T cells, and that this has a positive impact on patients’ survival, the same phenomenon has been subsequently validated by other groups in most solid tumors. Thus, how CD8+ T cell infiltration and engraftment is regulated in tumors has remained a key question in my lab. Furthermore, we know that response to PD-1 checkpoint blockade requires pre-existing T cell infiltration in tumors, which we commonly call a “hot” tumor microenvironment. Therefore, this question became all the more crucial to answer. My lab has worked for several years to decipher this mechanism.
What was the most surprising finding of this study for you?
1) How chemokines and their synergy can orchestrate cellular interactions in the tumor microenvironment for a tumor to reach an inflamed status and 2) how the tumor cells invent ways to evade this immune attack by silencing chemokines like CCL5.
What was the coolest thing you’ve learned (about) recently outside of the lab?
Emerging clinical studies show impressive pathological responses of resectable, but high-risk and aggressive cancers such as lung, glioblastoma and melanoma to neoadjuvant immune checkpoint blockade therapy. Tumor “hotness”, indicated by the infiltration of CD8+ T lymphocytes, remains an important predictor of the response to neoadjuvant immunotherapy and our study becomes very relevant for explaining the mechanisms that establish the inflammatory status of the tumors.



