Clinically, heterogeneous immune responses have been detected between patients, between tumors in the same patient, and even by disease site. Some tumor microenvironments (TME) are typically immune cold and respond poorly to immunotherapy, such as pancreatic ductal carcinoma (PDAC). However, there are some exceptions, which provides the hope that a mechanistic understanding of any differences will provide better therapeutic opportunities. Diamond et al. investigated differences in immune priming in various TMEs to assess site-specific tumor immune responses. Their results were recently published in Cancer Immunology Research.
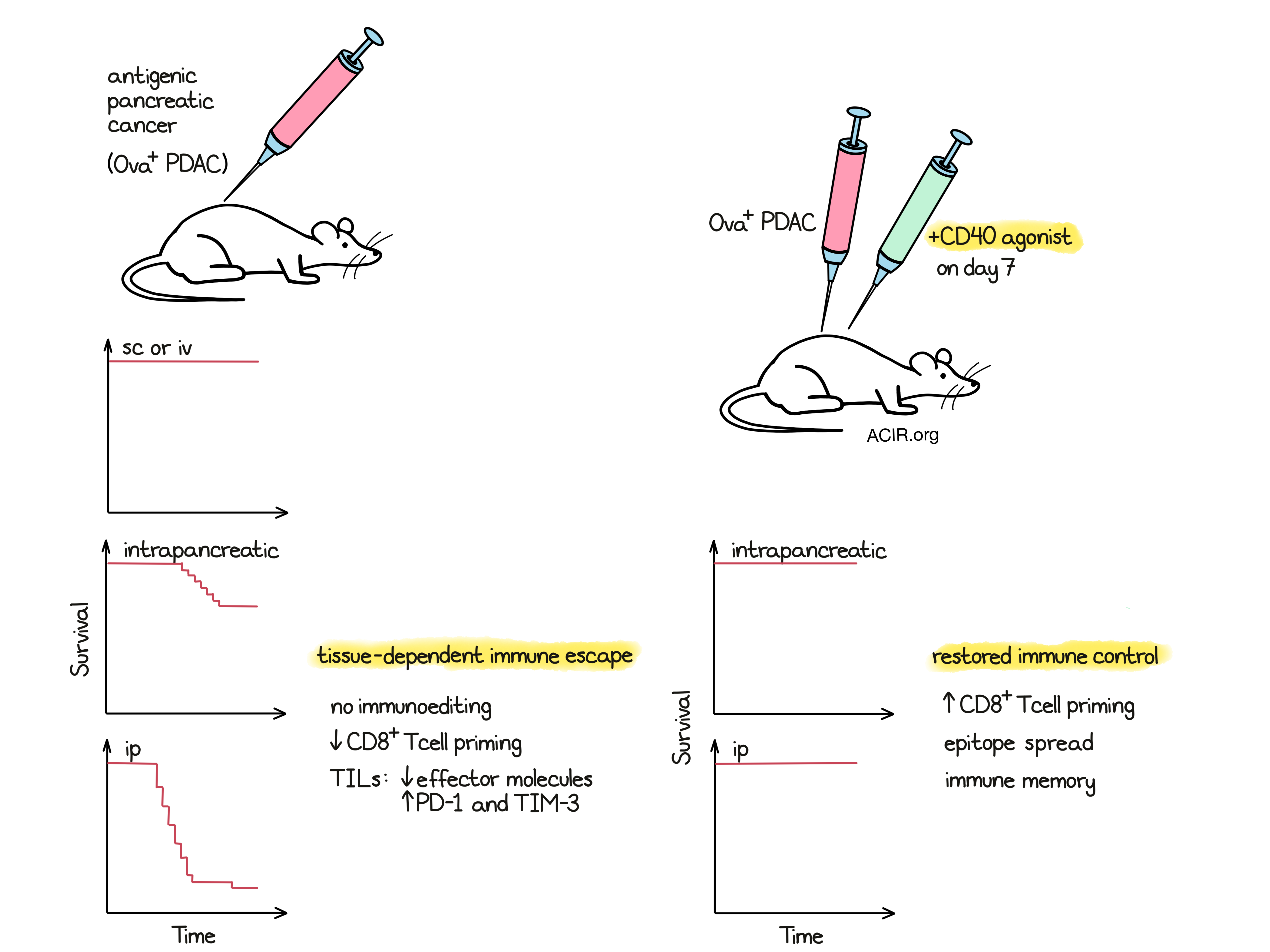
Diamond et al. made use of a murine PDAC cell line from a KRAS/tp53 GEMM tumor with enforced expression of ovalbumin (PDAC.Ova) as an immunogenic neoantigen. Both the parental PDAC and the PDAC.Ova lines grew morphologically similar tumors with a stable neoantigen load in immunodeficient Rag2-/- mice. However, when transplanted s.c. in immunocompetent mice, the PDAC.Ova line was rejected, which was dependent on CD8+ T cells, but not B or NK cells. The cells were also not rejected in mice with germline tolerance to ovalbumin (Act-mOVA). However, when cells were inoculated into the pancreas (orthotopic), 30% of mice experienced tumor growth. All of the mice grew tumors when CD8+ T cells were depleted, and rejection was not observed in ovalbumin-tolerant mice (Act-mOVA or orally tolerized). When 8x more cells were injected, pancreatic implantation resulted in tumor growth in 75% of mice, but only in 12% of mice that were s.c. injected. The researchers also assessed i.p. and i.v. injection in wild-type mice and found that the PDAC.Ova line grew in the peritoneum, but did not cause lung metastases after i.v. injection, in contrast to the parental line which was almost uniformly lethal at all sites. Therefore, the injection site affected the immune control of these neoantigen-expressing cells.
To determine whether the outgrowth of these tumors in the peritoneum and pancreas was due to a loss of neoantigens, the tumors were harvested from those sites and low-passage cell lines were generated. These tumor lines expressed ovalbumin, presented the Ova SIINFEKL peptide in the context of H-2Kb, and upregulated MHC-I upon IFNγ treatment. When cells were retransplanted s.c. into wild-type mice, tumors were rejected in most mice, but grew when CD8+ T cells were depleted. These data suggest that the immune escape was not due to immunoediting.
To further understand the differential immune response against the PDAC.Ova tumors, the researchers compared TIL and tumor-specific tetramer+ T cells (Ova tet+) in early (day 9) s.c. PDAC and PDAC.Ova tumors. The PDAC.Ova tumors had higher infiltration of CD8+ T cells and a decrease in CD11b+ myeloid cells. Ova tet+ T cells were found in spleens of wild-type mice that received s.c. tumor inoculation but were found at lower levels in mice that received i.p. or orthotopic injection of tumor cells. Few Ova tet+ CD8+ T cells were found in i.p. PDAC.Ova tumors. The CD8+ T cells found in s.c. PDAC.Ova tumors were of an effector phenotype, expressing activation markers and effector molecules, while the CD8+ TIL from i.p. and orthotopic PDAC.Ova tumors did not express effector molecules and had high levels of PD-1 and Tim3. These data suggest a reduced CD8+ T cell priming in the peritoneal and pancreatic microenvironments.
Given the critical role of cDCs in T cell priming, the authors then examined the infiltration of cDCs in tumors and tumor-draining lymph nodes (tdLN). There were no differences in total cDCs or cDC1s, or differences in expression of maturation markers in either the tumor or tdLN between the different tumor inoculation sites. However, since Batf3-dependent cDC1s play important roles in antigen cross-presentation, the T cell priming was assessed in Batf3-/- and CD40-/- mice. When cDC1s were absent (Batf3-/-), no CD8+ T cell priming occurred, while in the absence of CD40, the priming was reduced, but not absent. Tumors grew in Batf3-/- mice similarly to the CD8+ T cell-depleted mice. T cell priming and tumor growth were also affected in mice lacking secondary lymphoid tissue (lymphotoxin alpha-deficient mice).
Mice primed by s.c. tumor inoculation rejected high-dose orthotopic or i.p. tumors inoculated one week later, indicating that they developed immunological memory. However, no protective effect was found when the s.c. and i.p. injections were given simultaneously, or when the s.c. tumor was delivered one week after the i.p. tumor. Furthermore, i.p. and s.c. injections given at the same time reduced the number of infiltrating tetramer+ CD8+ TIL compared to mice injected with only a s.c. tumor, suggesting that the presence of the i.p. tumor inhibited antitumor activity.
Diamond et al. then hypothesized that enhancing APC functioning with CD40 agonist therapy might amplify T cell priming in the peritoneum and pancreas TMEs. Indeed, treating mice bearing orthotopic or i.p. PDAC.Ova tumors with a single dose of CD40 agonist 7 days after tumor implantation resulted in the rejection of tumors. In addition, there were higher levels of Ova tet+ cells in the spleens of mice bearing i.p. tumors treated with CD40 agonist, which was dependent on the presence of cDC1s. To explore whether checkpoint blockade could similarly boost the immune response, the researchers treated the mice with serial anti-PD-1 or serial anti-CTLA-4 therapy. Compared to agonist CD40 therapy, PD-1 blockade had a limited effect on tumor rejection, and CTLA-4 treatment resulted in partial protection, consistent with the known effect of anti-CTLA-4 on T cell priming.
Finally, to determine whether CD40 agonism resulted in a broadening of the specificity of T cell responses, the researchers inoculated Ova+ and Ova- tumor cell mixes at the same site, or Ova+ and Ova- tumors on opposite flanks. Treatment with the CD40 agonist resulted in complete rejection of tumors, which was dependent on T cells and cDC1s. Priming mice with s.c. PDAC.Ova and rechallenging with PDAC also resulted in complete control of the tumors. These data suggest CD40 agonism results in epitope spreading and induction of memory responses.
Taken together, these results suggest that the immune response differences seen in various tumor microenvironments might be the result of site-specific differences in T cell priming, a defect that could be overcome by APC stimulation through CD40 agonism. If these results can be confirmed in patients, this may add a new tool to help cold immune environments turn hot and increase immunotherapy efficacy.
Write-up by Maartje Wouters, image by Ute Burkhardt



