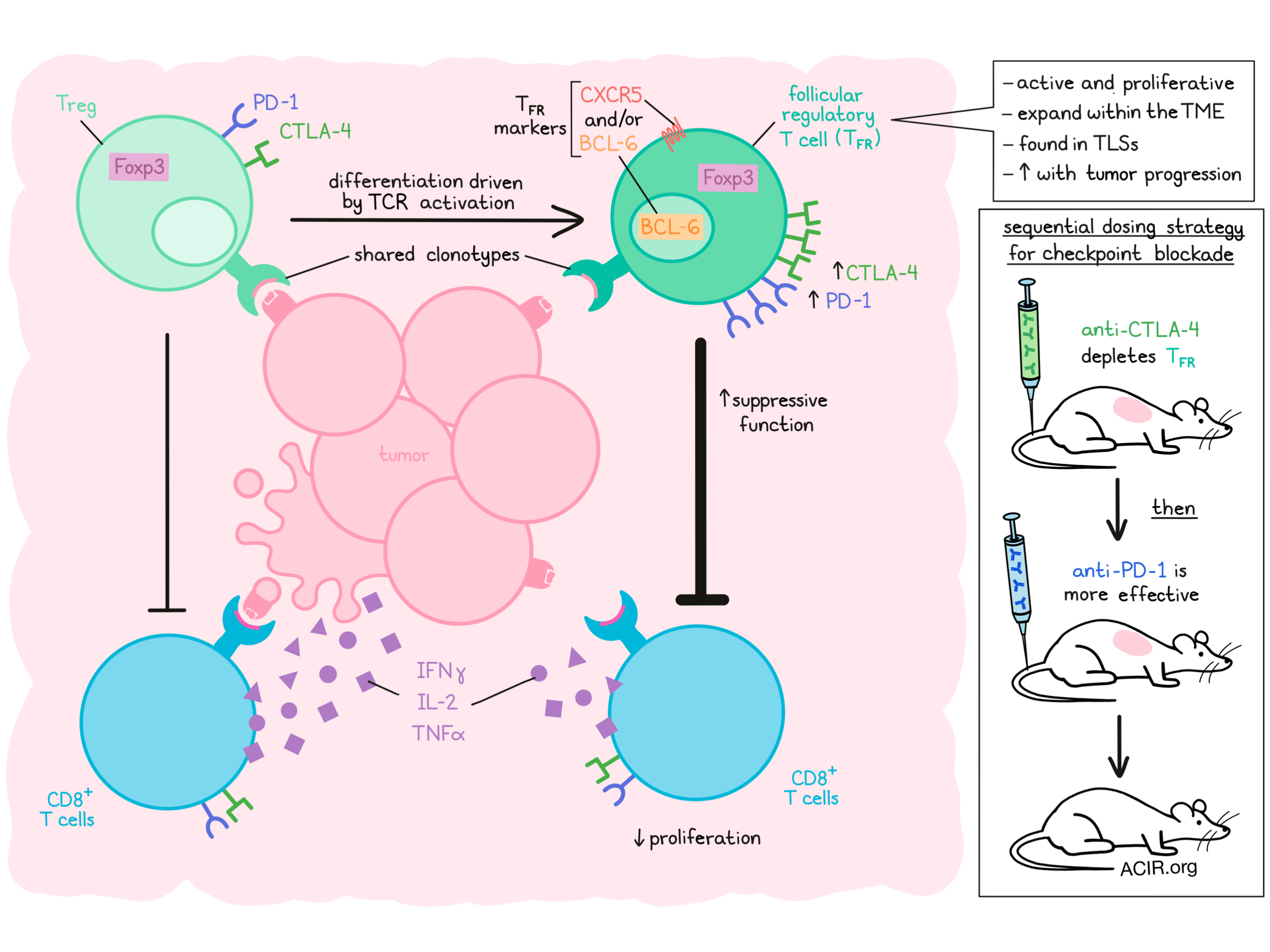
Regulatory T cells (Treg) can differentiate into follicular regulatory T cells (TFR) that have important roles in secondary lymphoid organs and may have higher suppressive activity. However, their role in antitumor immunity has not been elucidated. Eschweiler et al. investigated the role of TFR in cancer, analyzing their function, characteristics, and effects on the efficacy of checkpoint inhibition using RNAseq of human cancer cohorts and mouse models. Their results were recently reported in Nature Immunology.
The researchers began by analyzing nine published single-cell RNAseq datasets to assess CD4+ TIL in six human cancer types. Between 5 and 55% of the evaluated CD4+ TIL were FOXP3-expressing Tregs, and of these, 5-30% also expressed BCL6 and/or CXCR5, indicative of follicular lineage TFR cells. The presence of TFR was confirmed in tumor samples from patients with non-small lung cancer (NSCLC), where they were mainly found in tertiary lymphoid structures (TLS). TFR also expressed CD25 and ICOS and expressed the highest levels of CTLA-4 and PD-1 of all TIL.
Murine TFR obtained by immunizing mice with ovalbumin (OVA) and adjuvant had similar transcriptomes as Tregs, such as genes encoding heightened suppressive capacity, and were associated with CD8+ T cell dysfunction and survival, suggesting these cells have a suppressive potential. Performing bulk RNAseq on enriched subsets of Tregs and TFR TILs from human NSCLC revealed a cluster that correlated with the TFR phenotype. This cluster also expressed BCL6 and FOXP3, and was enriched for genes involved in the cell cycle, translational and transcriptional activity, TCF-1 expression, and mTOR signaling. These data suggest TFR are active and proliferative in the tumor microenvironment (TME).
The researchers then hypothesized that tumor antigen recognition might trigger conversion from Tregs to TFR within the TME. To analyze this, they sorted CD4+ and CD8+ populations from the tumor tissue and tumor-infiltrated lymph nodes from two patients with head and neck squamous cell carcinoma and subjected these cells to single-cell RNAseq and TCR sequencing. Two distinct FOXP3-expressing CD4+ clusters were detected, which had distinct transcriptomic signatures indicative of TFR and Tregs. TFR expressed higher levels of transcripts linked to metabolism, cell activation, co-stimulation, TFR function, suppressive capacity, and cell cycle than Tregs.
There were also shared clonotypes between Treg and TFR, but TFR were more clonally expanded. A trajectory analysis suggested that conversion from Treg to TFR happened in the tumor. In a separate large single-cell RNAseq dataset of CD4+ TIL, most clonally expanded clonotypes of TFR were shared with Tregs. The Tregs that shared clonotypes with TFR expressed 4-1BB, suggestive of recent TCR activation. Genes linked to cell activation, co-stimulation, and suppression were expressed at higher levels in the 4-1BB+ TCR-sharing Tregs, and in the clonally expanded TFR. Additionally, TFR expressed lower levels of CCR7 and S1PR1, suggesting these cells have tissue-resident characteristics.
Eschweiler et al. then used the B16F10 melanoma and the MC38 colorectal tumor model system to assess tumoral TFR function. Similar to human cells, intratumoral TFR had increased Ki67, TCF-1, and 4-1BB expression compared to Tregs. In vitro and in vivo experiments showed that these cells were more suppressive than Tregs, inhibiting proliferation and reducing the secretion of IFNγ, IL-2, and TNFα by CD8+ T cells. Transferring OT-I T cells alone or with Tregs or TFR in B16F10-OVA tumor-bearing RAG1-knockout mice showed that TFR inhibited OT-I tumor cell killing, with only a limited effect of Tregs, further confirming the highly suppressive function of TFR.
Single-cell RNAseq of barcoded Foxp3+CD4+ T cells from B16F10-OVA tumor-bearing mice, at day 11 and day 18 of tumor growth, revealed five clusters. Only one cluster was enriched on day 18, and this cluster was enriched for genes previously linked to TFR, suggesting these cells increase in the tumor over time. Constructing a developmental path with cell-trajectory analysis revealed that TFR cells develop from a cluster of cells with naive, recirculating Treg features. To confirm this further, Eschweiler et al. conducted a time-course experiment, which showed that intratumoral TFR, but not Tregs, increased with tumor progression.
Like their human counterparts, murine TFR expressed high levels of CTLA-4 and PD-1. Therefore, these cells might respond to checkpoint inhibition therapy. Indeed, anti-PD-1 treatment increased the number of TFR in MC38 and B16F10 tumors, potentially opposing the benefit of PD-1 blockade. Re-analysis of published single-cell RNAseq data from patients who had received PD-1 blockade therapy suggested that TFR were enriched for T cell activation and costimulation transcripts after therapy. To assess the specific inhibitory effects of TFR, apart from any Treg inhibition, the authors used a genetic knockdown system to selectively deplete TFR, and found that knockdown increased treatment efficacy.
To further validate the role of TFR in tumor development and its effects on anti-PD-1 treatment, the researchers used the B16F10 tumor model, which is known to not respond to anti-PD-1 therapy, and modified it to be TFR-deficient (Foxp3cre- Bcl6fl/fl). Without TFR, tumor growth was less robust, and anti-PD-1 therapy further increased tumor control, suggesting TFR indeed decrease the efficacy of anti-PD-1 therapy. CD8+ T cells in the lymph nodes of these TFR-deficient mice had higher expression of granzyme B, suggesting TFR may inhibit CD8+ T cell function in the lymph nodes. A competition assay showed that BCL-6 was required for the tissue persistence of Foxp3+ cells in the TME.
Given its inhibiting effects on anti-PD-1 therapy, the researchers hypothesized (1) that depletion of TFR before treatment might enhance efficacy and (2) that given the high expression levels of CTLA-4 on these cells, blockade of CTLA-4 might deplete TFR. Monotherapy of anti-CTLA-4 indeed depleted TFR preferentially over Tregs, and sequential treatment with anti-CTLA-4 followed by anti-PD-1 resulted in a reduction in tumor volume compared to either monotherapy, and elevated the frequency of CD8+ TIL and granzyme B+ CD8+ and CD4+ T cells in the B16F10-OVA model.
Finally, retrospective analysis of survival of patients with unresectable melanoma treated with either anti-CTLA-4 or anti-PD-1 monotherapy, simultaneous combination therapy, or sequential anti-CTLA-4 and anti-PD-1 therapy showed that patients in the sequential treatment group had the best outcome. In addition, a cohort of patients with melanoma showed that those who had poor survival and did not respond to anti-PD-1 treatment had more FOXP3+BCL-6+CD4+ TIL, indicative of TFR.
Together, these data from Eschweiler et al. suggest a significant role of TFR in cancer, the presence of TFR in TLSs could potentially predict which patients to treat with sequential checkpoint inhibition therapy, and that TFR may be a new target for immunotherapy.
Write-up by Maartje Wouters, image by Lauren Hitchings



