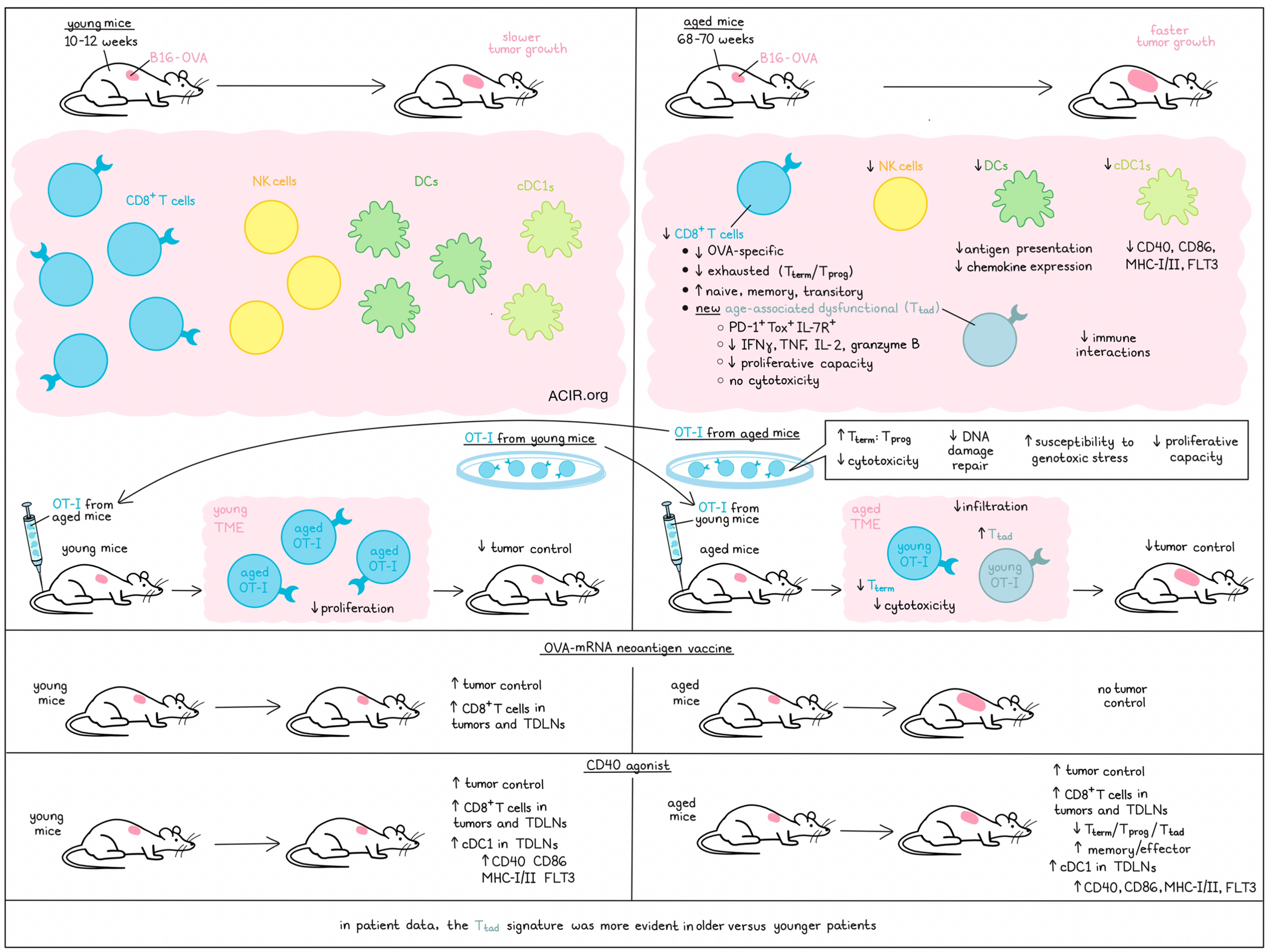
Aging is known to induce immune dysfunction in cancer, but the mechanisms behind it remain poorly understood. In a recent Nature Immunology publication, Chen et al. assessed tumor-infiltrating T cells and the tumor microenvironment (TME) of young and aged mice to determine how age-related dysfunction relates to chronic stimulation-induced T cell exhaustion.
To evaluate the effects of aging on the antitumor immune response, OVA-expressing B16 melanoma (B16-OVA) tumors were implanted in C57BL/6J mice of various ages. In mice aged 68-70 weeks (aged mice) tumor growth was faster than in 10–12-week-old mice (young mice). Further, the aged mice had fewer tumor-infiltrating CD8+ T cells and OVA-specific CD8+ T cells.
scRNAseq of the tumor-infiltrating CD8+ T cells revealed several populations of Pdcd1- and Tox-expressing exhausted cells, progenitor-like exhausted Tcf7- and Slamf6-expressing cells (Tprog), and terminally exhausted cells (Tterm), characterized by Havcr2 and Entpd1 expression. In the aged TME, exhausted T cells were depleted, while less differentiated subsets were increased, including naive-like (Tnaiv), memory-like (Tmem-like) and transitory (Ttran) CD8+ T cells. Further, there was a cluster only expressed in the aged TME characterized as PD-1+Tox+IL-7R+CD8+ T cells, which did not express Tcf7, Slamf6, or Havcr2. The researchers named this subset tumor-infiltrating age-associated dysfunctional T cells (Ttad). In the aged TME, there was increased Ttad cell formation, including within the OVA-specific subset, and a decrease in Tterm cells. The aged tumoral CD8+ T cells produced less IFNγ, TNF, IL-2, and granzyme B, and had lower proliferative capacity.
To determine whether these age-related differences could be explained by defects in baseline T cells in healthy aged mice, the researchers transferred OT-1 cells from young mice (OT-1y>y) and OT-1 cells from aged mice (OT-1a>y) into young tumor-bearing mice, and profiled the tumor-infiltrating cells. The aged OT-1a>y had a higher ratio of Tterm to Tprog cells than the young OT-1y>y and the ratio of Tterm to Tprog cells skewed further over time during tumor progression. Before transfer, at baseline, many of the characteristics of the aged population were already present. Additionally, the aged OT-1 CD8+ T cells had reduced cytotoxicity in vitro, and OT-1a>y did not induce tumor control in young mice, in contrast to the young cells.
To assess how these defects are encoded in the aged T cells, chromatin accessibility changes were profiled between young and aged CD8+ T cells at baseline and after transfer in the TME. DNA damage/repair and replicative senescence signatures were differentially regulated in aged OT-1a>y Tterm cells, suggesting these cells might be more susceptible to genotoxic stress. Exposing the cells to UV irradiation confirmed that the aged cells upregulated the DNA damage markers γH2AX and p21 more rapidly and to higher levels. In vivo, the aged population had reduced proliferation in young mice, while young OT-1 that were transferred into aged mice showed no difference in proliferation to those transferred into young mice, even though the endogenous T cells in the TME had lower proliferation capacity.
The researchers did not detect the Ttad population in tumors of young mice transferred with aged OT-1. However, when young OT-1 were transferred into aged mice, there was Ttad formation and a loss of Tterm cells. Therefore, the formation of Ttad was not an intrinsic CD8+ T cell characteristic, but required signals from the aged TME. To further assess this, young OT-1 were transferred into young (OT-1y>y) and aged (OT-1y>a) mice with B16-OVA tumors. The transfer of the young OT-1 did not induce tumor control in the aged mice, and fewer transferred cells ended up in the tumor.
Chen et al. isolated intratumoral OVA-tetramer+CD8+ (Tet+CD8+) T cells and OT-1 populations from tumors in young and aged mice and performed coculture experiments. This showed that young and aged Tprog cells had low cytotoxicity, young Tterm had higher cytotoxicity than aged Tterm cells, and Ttad cells had almost no cytotoxic activity. When Tet+CD8+ cell populations were transferred into young mice, those receiving young Tprog cells had better tumor control, while there was no benefit from Ttad cells.
To determine whether Ttad cells could also be detected in human cancers, published pan-cancer scRNAseq datasets were assessed, and a similar cluster of PD-1+IL-7R+CD8+ T cells not expressing SLAMF6 or Tim3 was found to be enriched for the transcriptional signature of the mouse Ttad. This cluster was more abundantly present in older than in younger patients.
Profiling the immune populations in aged murine tumors, the researchers found that only CD8+ T cell, NK cell, and DC levels were lower than in young tumors. The aged TME had lower expression levels of antigen presentation and chemokine genes. Aged DCs downregulated antigen presentation molecules B2m and Tap1, and upregulated Apoe, which inhibits antigen presentation. There were fewer interactions between NK cells, cDC1, and CD8+ T cells in the aged TME. Aging tumors and tumor-draining lymph nodes (TDLNs) had fewer cDC1s, which expressed lower levels of CD40, CD86, MHC-I/II, and FLT3.
Finally, the effects of an OVA-mRNA neoantigen vaccine, optimized using lipid nanoparticles, were assessed in young and aged mice. While the vaccine induced tumor control and increased CD8+ T cell tumor and TDLN infiltration in young mice, there were no effects in aged mice. Since CD40 agonists can increase myeloid cells and their function in tumors, the researchers alternatively evaluated whether activating the cDC1 population in the aged TME could improve CD8+ T cell antitumor responses. Treatment with an agonist anti-CD40 antibody improved tumor control in both young and aged mice, and it increased cDC1s in the TDLN, and CD8+ T cells in the tumor and TDLN. Treatment induced upregulation of CD40, CD86, MHC-I/II, and FLT3 on aged cDC1s and decreased the Tprog, Tterm, and Ttad subsets, while increasing memory- and effector-like subsets.
These data suggest that aging-related unfavorable antitumor responses are linked to a newly-identified T cell subset, which appears due to reduced interactions with cDC1s in the TME. These findings have important implications. Therapies that enhance the interactions between cDC1s and CD8+ T cells may improve therapeutic efficacy in older patients.
Write-up by Maartje Wouters, image by Lauren Hitchings.



