
The ACIR team attended the CRI-ENCI Ninth International Cancer Immunotherapy Conference (CICON) 2025 in Utrecht, The Netherlands. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Keynote address
Michel Sadelain
William B. Coley Award Lecture
Aviv Regev
Tumor immune microenvironment
Dmitry Gabrilovich
Tanja de Gruijl
Pascale Zwicky
T cell therapies
Cedrik Britten
Laurie Menger
Alexandra Reinbrecht
Antigen discovery and cancer antigen vaccination
Ton Schumacher
Rienk Offringa
Michal Bassani-Sternberg
Vinod Balachandran
Neoadjuvant immunotherapy
Tina Cascone
Overcoming immunotherapy resistance
Catherine Sautès-Fridman
Juan Osorio
Michael Curran
In situ cancer vaccination
Arthur Krieg
Keynote Address
Principles of CAR design - Michel Sadelain, Columbia University, USA
In his keynote lecture, Michel Sadelain traced the history of CAR T cells, including the cloning of the CD3ζ-chain in 1991, Zelig Eshhar’s T-body construct that linked the CD3ζ-chain with an scFv, and his own work on introducing genes into primary T cells to reflect natural T cell biology. By including the CD28 costimulatory domain in CD3ζ-chain-fusion proteins (now called 2nd generation CARs), it became possible to redirect the cytotoxicity and expand engineered primary T cells upon repeated antigen exposure. Having very rationally chosen CD19 as a target a decade earlier, in 2003, Sadelain and his team reported the first efficacy of human CD19-targeted CAR T cells in immunodeficient mice with B-cell malignancies. By 2017, the first two CD19 CAR T cell therapies, Yescarta and Kymriah, were approved by the FDA for the treatment of relapsed refractory B cell malignancies, leading to high complete remission rates (five CD19- and two BCMA-directed CAR T cell products are now approved). Anti-CD19 CAR T cells have also shown remarkable responses in systemic lupus erythematosus, and are being tested for other autoimmune diseases. Receptor sensitivity to detect low-abundance targets and T cell exhaustion need to be addressed to advance this immunotherapy. CARs form a weak synapse, and lower target levels at relapse highlight the need for more sensitive receptors. Based on the high sensitivity of target detection by the multi-component T cell receptor (TCR), Sadelain and his colleagues created a “HIT” receptor where the VL and VH of the CAR have been transposed to the constant regions of a TCR, making the receptor HLA-independent, but CD3-dependent. As an example of enhanced sensitivity, the anti-CD70 HIT receptor containing a costimulatory domain achieved striking and durable responses in patient-derived xenograft models of renal cell carcinoma where CD70 expression was dramatically down-regulated, almost negative, due to epigenetic regulation, and CD70-directed CAR T cells were strikingly ineffective. HIT receptors are estimated to require 5-10 fold fewer target molecules than CD28ζ or 4-1BBζ CARs. As another approach to enhance sensitivity of CAR T cells, Sadelain explored "IF-BETTER" logic-gates to engage two antigens, combining a CAR for a primary, low-expressed antigen with a chimeric costimulatory receptor (not harboing the CD3ζ-chain) as helper molecule for a secondary antigen. This approach enhanced sensitivity in models of AML, allowing effective killing of tumor cells expressing suboptimal levels of ADGRE2 and the secondary antigen CLEC12A. These ADCLEC.syn1 CAR T cells are now being evaluated in a phase I clinical trial with initial encouraging results. To address the issue of functional persistence of CAR T cells, Sadelain and his team have calibrated CAR signaling strength by modifying the ITAM motifs in the CD3ζ chain. The "1XX" CAR, with only one active ITAM, showed improved persistence and efficacy in CAR stress cancer models and in clinical trials, achieving durable remissions at much lower doses and with reduced toxicity compared to current commercial products. Finally, inspired by the observation that thymic T cells containing the re-arranged TCRβ with the unrearranged pre-TCRα chain underwent dramatic expansion, Sadelain incorporated elements of the pre-TCRα cytoplasmic domain into CARs. The "1A" CAR boosted T cell accumulation, durability, and therapeutic efficacy by increasing YBX1 phosphorylation, thereby enhancing global mRNA translation.
William B. Coley Award Lecture
William B. Coley Award Lecture: From Cell Atlases to Medicines with AI - Aviv Regev, Genentech, USA
Aviv Regev, the recipient of the prestigious William B. Coley Award, used her talk to educate and excite her audience about the coming involvement of artificial intelligence (AI) in science, and particularly cancer immunotherapy – an opportunity that her pioneering work in collecting and analyzing large collections of data derived from single cells has significantly contributed to. The fundamental problem of cancer is enormously complex, but single-cell genomics technology has now emerged as a tremendously useful tool to tease apart and describe the cellular components, and spatial technology is rapidly determining their spatial arrangement. With more than 200 million cells – from normal tissues, pathological tissues, and tumors – now characterized and annotated at the single-cell level, more sophisticated questions can be addressed, like 'Where can I find the same kind of macrophage I saw in lung fibrosis?", without that cell type ever having been uniquely annotated before. This example illustrates a new principle enabled by big data approaches, even with messy data. Quantity of data can become a quality itself, just like in-depth, one-feature-at-a-time analysis by an investigator builds quality. AI “learns” what contributes to various cell properties by training on these large datasets and identifying patterns, which we use to characterize individual cell types, define cell differentiation trajectories, understand broad gene programs active in different cell types, and more. As a first example, characterization of T cells/tumor cells in “hot” and “cold” tumors identified an “exclusion” signature in tumor cells in areas devoid of T cells, and this signature was associated with poor response to immune checkpoint blockade (ICB). Analysis of a large dataset on the transcriptional effects of various drugs on human cell lines in vitro predicted that CDK4/6 cell cycle regulators drove this exclusion program. This led to the hypothesis, subsequently shown to be correct, that inhibitors of CDK4/6 would reverse exclusion and re-install sensitivity to ICB. Recognizing that much biology happens as communication between cells, Regev next described DIALOGUE, an algorithm to uncover co-varying gene programs identifying cell:cell interactions and effects. In most cases, data and AI will not be enough to get to the desired answers, and Regev introduced the concept of “Lab in a Loop” – an iterative process of learning from available data, systematically collecting new, related data in the lab, and repeating the AI/lab cycle. One particularly useful lab approach is large perturbation screens (Perturb-Seq) which can interrogate pooled perturbations with single-cell readouts to deconvolute the effects. Bar-coded antibody staining (CITEseq) can be added on to provide important surface protein level expression information. Finally, matrix algebra-based algorithms can be used to define higher level, co-regulated control programs and their antecedent drivers. This approach can be used to determine the impact of key mutations in cancer-driving genes, including, for example, how such mutations alter the composition of the tumor microenvironment or tumor histology. Regev went on to describe how the algorithms Paired and Unpaired SCHAF can be trained on complete single-cell transcriptomic, spatial transcriptomic, and histological information from a set of tumors, which can then be used to infer the transcriptomic and spatial data for a new tumor based only on histological information. Similarly, combining PerturbSeq (to determine cell phenotype) and PerturbView, a highly plex methodology based on optical pooled screening technology, revealed concordant perturbed regulatory maps relating expression and imaging phenotypes, allowing inferences for unseen perturbations. Validation data on breast cancer samples and for macrophage signaling was very encouraging, opening up endless possibilities to understand cell and tissue circuits. Still, such approaches are only touching on the enormous complexity of tumors, and Regev went on to explain how small “compressed” experiments and iterative active learning leverage this basic perturbation information and can be used to efficiently fill in the gap. The next challenge is how to use such modeling and iterative loops to discover the best molecules to test in clinic, even on an individual patient level, such as for a personal neoantigen cancer vaccine, which has been done with encouraging results.
Tumor immune microenvironment
Therapeutic targeting of myeloid cells - Dmitry Gabrilovich, AstraZeneca, USA
Myeloid cells are crucial in the suppression of immune responses to cancer, but targeting these cells has shown limited clinical success despite extensive research. Over the recent years, the understanding of extensive myeloid cell heterogeneity has shifted from a multi-subset concept, which presented challenges due to transcriptional flexibility and varied functions in different cancers and disease stages, to a concept based on functional features. This new concept proposes that myeloid cells in cancer exist in a limited number of functional states, often with significantly overlapping transcriptional patterns, transcending those transcriptional profiles. Dmitry Gabrilovich and others have identified two major functional states. The classical activation state is driven by short and strong signals that are derived from pathogens. In cancer, however, pathological activation of myeloid cells occurs through weak activation signals from growth factors and cytokines that are present over a long period of time, leading to immune suppression. Such a functional dichotomy may extend beyond macrophage-related myeloid cells to neutrophils and dendritic cells. Clinical data confirms a link between high abundance of myeloid-derived suppressor cells (MDSCs) to negative prognostic impact and immunotherapy resistance. However, successful targeting of myeloid cells is hampered by two major roadblocks: cellular redundancy (different myeloid lineages share similar functional activity) and molecular redundancy (converging alternate intracellular pathways). Overcoming these challenges requires broad, impactful targeting, not overly specific approaches. Clinical trials targeting the DNA damage response with the ATR inhibitor ceralasertib and anti-PD-L1 showed promise, with a 14% response rate and improved progression-free and overall survival in randomized settings in patients with NSCLC who had progressed on anti-PD-(L)1-based therapy. Interestingly, the antitumoral effect of ceralasertib was mediated by type I IFN and CD8+ T cells. Cerlasertib led to the depletion of M-MDSCs and TAMs, without affecting DC numbers. Although polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) numbers were also not affected, upregulation of the IFN-I pathway in these cells was associated with an inactivation of their suppressive activity. At the same time, DCs were activated via IFN-I signaling. Through this multifunctional effect on cells within the tumor microenvironment, ceralasertib targeted a cellular redundancy roadblock. Garbrilovich then presented a promising approach targeting the problem of molecular redundancy in myeloid cells. He and his team focused on ferroptosis, a form of regulated cell death. This process is initiated by the disruption of regulatory redox mechanisms, leading to extensive peroxidation of polyunsaturated phospholipids and multiple attempts to induce ferroptosis of tumor cells are under investigation. Gabrilovich found that neutrophils in tumors showed a signature of ferroptosis, which induced a strong immunosuppressive state, and that the immunosuppressive state of PMN-MDSCs isolated from tumors could be reversed with inhibitors of ferroptosis. Moreover, neutrophils are highly resistant to ferroptosis compared to other immune cells or to tumor cells. They achieve this primarily by releasing extracellular vesicles containing oxidized lipids, essentially "spitting out" the damaging elements. This mechanism allows neutrophils to survive longer in the tumor microenvironment while simultaneously delivering pro-tumorigenic factors, benefiting the tumor. In conclusion, targeting myeloid cells is possible, but success requires addressing redundancy and focusing on fundamental mechanisms rather than individual biochemical pathways.
Convert or die: going medieval on TAMs - Tanja de Gruijl, Amsterdam UMC
Tanja de Gruijl began by pointing to her provocative presentation title comparing the players and questions in the Spanish Inquisition to the roles of tumor-associated macrophages (TAMs) in the TME, with a hint that the TAMs are not always the bad guys who need to be converted. The cancer immunity cycle points to the critical role that dendritic cells (DCs) play in the TME and associated lymphoid tissues to induce and maintain T cell immunity. As tumors have evolved to disrupt this process, primarily through a variety of soluble factors, understanding the composition and dynamics of the factor-producing cells in the TME is important. Myeloid cells (CD11c+, HLA-DR+) make up the dominant proportion of non-tumor cells in the TME (excluding neutrophils, which are not isolatable from frozen tissue samples), dwarfing the DC population, and these myeloid cells are predominantly monocyte-derived (CD14+). Monocytes can differentiate into DC-like cells, but predominantly differentiate into TAMs, with a high abundance of DC-like cells being associated with favorable prognosis, and a high abundance of TAMs being associated with poor prognosis. De Gruijl identified optimizing the DC/TAM balance as a potential way to improve outcomes, using other innate cells to affect TAM biology. The first approach was with NK cells. Beyond tumor cell killing, NK cells can recruit and activate DCs. Using cord blood-derived allogeneic human NK cells in co-culture experiments with human tumors, de Gruijl showed that these NK cells reduced the CD14+ TAM population and increased mature CD1c+ DCs, which could be further activated with R848. These effects were independent of the presence of tumor cells. De Gruijl then described an approach using NKT cells, which have an invariant TCR that recognizes lipid antigen-bound CD1d. A bispecific antibody was developed that (1) bound to tumor-expressed CD1d and converted it to an antigen recognizable by cytolytic type 1 NKT cells, and (2) bound to Vδ2 on γδT cells to colocalize these cytolytic cells. Many tumors express CD1d, frequently at levels much higher than conventional class I MHC. Co-culture of human CD1d+ tumor cells with human type 1 NKT cells, human γδT cells, and the bispecific antibody led to degranulation of both effector cell types, and resulted in tumor killing. Importantly, this bispecific and γδT cell combination also eliminated CD14high and CD1d+CD163+ macrophages, remodeling the TME and enhancing the pro-inflammatory cytokine profile. The supernatant from these remodeled cocultures was able to abrogate the differentiation of monocytes to M2-like macrophages, and instead increased DC-like cells. Finally, de Gruijl described an anti-PD-L1xVδ2 bispecific engager that was capable of enhancing lysis of human tumor cells by blocking T cell inhibition and redirecting γδT cells. The bispecific increased the infiltration of tumor spheroids with Vγ9Vδ2 T cells and induced lysis of tumor cells. In cultures of melanoma cell suspensions, the bispecific led to a significant reduction of TAMs, and instead induced signals of more mature DCs. De Gruijl hypothesized that DCs are not eliminated by Vδ2-directed bispecifics, even though they can express PD-L1 and CD1d, due to their higher resistance to apoptosis (high expression of the apoptosis inhibitor PI-9).
A tumor-associated macrophage-targeted immunocytokine leveraging T and NK cell synergy for potent cancer immunotherapy - Pascale Zwicky, Weizmann Institute of Science, Israel
Focusing on eliminating suppressive tumor-associated macrophages (TAMs), Pascale Zwicky began by targeting TREM2, a TAM surface molecule critical to TAM function. Although treatment with an anti-TREM2 led to phenotypic changes in TAMs, the antitumor effects in the MC38 model were limited. Based on spatial transcriptomic data across several tumor types, T cells and TAMs showed the closest spatial proximity. Receptor-ligand interactome analysis also showed strong predicted interactions between TAMs and T cells, most of which were T cell-suppressive. This suggested testing an anti-TREM2-IL-2 immunocytokine as a potential way to capitalize on this proximity, but this molecule was highly toxic, with signs of a cytokine storm. This problem was solved by adding an IL-2 mask connected with a TAM-specific protease (MMP14) cleavable linker, creating a myeloid/tumor microenvironment-targeted immunocytokine (MiTE). The MiTE-144 configuration was optimal, and led to significantly improved tumor control in the MC38 model, which was enhanced with anti-CTLA-4. Single-cell sequencing of tumors showed TAMs were increased in hypoxic signatures and more inflammatory monocyte-derived macrophages, while T cells were more stem-like and less exhausted, and NK cells were more cytotoxic, and more proliferative. Treatment of human patient-derived RCC tumor fragments with MiTE-144 and anti-PD-1 showed encouraging enhanced CD8+ T cell and NK cell effects, and a reduction in different TAM populations.
T cell therapies
PRAME-TCR: From preclinics to clinics - Liane Preußner, Immatics Biotechnologies, Germany
High PRAME expression is found in many cancers, including melanoma and ovarian cancer, and is nearly absent in normal tissues, making it an ideal therapeutic target. PRAME is an intracellular protein that enhances tumor survival and proliferation. PRAME expression is homogenous in tumor tissue and associated with poor prognosis. A parental TCR recognizing a PRAME epitope presented on HLA-A*02:01 was cloned from a healthy donor and optimized based on structural information to stabilize the interaction between the α and β variable domains, which also enhanced its binding affinity and activation, even against cells with low PRAME expression levels. Liane Preußner reported on the clinical results observed with PRAME-directed TCR T cell therapy (Anzu-cel) for the treatment of advanced and/or metastatic PRAME-expressing HLA-A*02:01-positive solid tumors in a phase 1 trial. Manufacturing of Anzu-cel took 14 days, followed by 7 days for quality control, achieving a 95% success rate. Patients received lymphodepleting chemotherapy (fludarabine and cyclophosphamide) followed by the TCR T cells and low-dose s.c. IL-2 over 10 days. A total of 74 patients were treated, most of whom had received multiple prior treatments and had large tumor volumes. The safety profile was favorable. Cytopenias were common, but not complicated by infection. CRS was seen in nearly all patients, mostly of low grade, with higher-grade CRS correlating with better clinical response. ICANS was less frequent, and mostly grade 1/2. Tolerability in the melanoma subset (n=33) was consistent with the full safety set, even though the patients with melanoma had a higher average dose. The confirmed objective response rate in patients with melanoma was 56%, with 50% in cutaneous melanoma and 67% in uveal melanoma. Median duration of response was currently at 12 months for all melanoma, and not yet reached for cutaneous melanoma, with some patients responding for 2-3 years now. TCR T cell expansion and persistence correlated with clinical responses. The median progression-free survival (PFS) and overall survival (OS) were 6.0 and 13.9 months, respectively, for patients with cutaneous melanoma, and 8.5 and 16.2 months, respectively, for patients with uveal melanoma. This encouraging data led Anzu-cel to earn the FDA Regenerative Medicine Advanced Therapy (RMAT) designation. A pivotal trial was recently started in advanced cutaneous melanoma, focusing on patients who have progressed on checkpoint inhibition. For this trial, PRAME testing was no longer required due to high prevalence, and patients were randomized to Anzu-cel or investigator’s choice. For other PRAME-expressing cancers, Preußner and her team developed a second-generation CD4/CD8 T cell product (IMA203CD8) containing PRAME TCR-engineered CD8+ and CD4+ T cells by adding the CD8αβ chain to the construct, allowing CD4 T cells to also be activated. In a phase 1 trial of this new product, safety was manageable, with cytopenias and CRS. One grade 4 CRS, but no treatment-related deaths, were observed, and two dose-limiting toxicities led to protocol amendment. Encouraging efficacy data includes a 41% overall response rate, with deep and durable responses. One patient with cutaneous melanoma and one with synovial sarcoma achieved complete responses. Lower doses were sufficient for clinical response with the second-generation product, offering an opportunity to treat cancers with both high- and medium-level PRAME expression.
Systematic identification of targets improving T cell therapies - Laurie Menger, Institut Gustave Roussy, France
Laurie Menger sought to enable and enhance allogeneic T cell therapies – which have multiple advantages over conventional autologous T cells, including faster utilization and lower cost – by identifying genes involved in resistance of allogeneic T cells to host T and NK cell attack (allogenic rejection). A genome-wide CRISPR-Cas9 knockout library was inserted into C57BL/6 murine CD4+ T cells, and cells that survived in vivo passage in an allogeneic BALB/c host were recovered to identify potential targets. The Fas Cell Surface Death Receptor (Fas) and Beta-2-microglobulin (B2m) quickly emerged as targets that led to higher T cell recovery. Cell depletion experiments demonstrated that allogeneic rejection depended on both NK and CD8+ T cells, with the latter being more significant. Deletion of NK cells had a minimal impact on resistance for Fas or B2m knockouts (KOs), while with the depletion of CD8+ T cells Fas KO cells outperformed B2m KO in terms of resistance. The stronger effect of CD8+ T cell depletion (which retains allo-NK cells) more closely mimicked the clinical situation, with NK cells being the first to repopulate after conditioning lymphodepletion. This led to a focus on the differential improvement mediated by Fas KO compared to B2m KO. Moving to the human system, Menger knocked out TCRs and either Fas or B2m, and observed in vitro that TCR/Fas KO cells were more resistant to allogeneic T cell attack. KO of both Fas and B2m did not further improve resistance compared to TCR/Fas KO cells. In vivo, TCR/Fas and TCR/B2m KO in CAR T cells targeting NALM6 tumors led to similar persistence in the face of allogeneic T cell challenge, but survival was enhanced by Fas KO, but not by B2m KO. Thus, Fas KO reduces the sensitivity of the CAR T cells to allo-NK rejection, and thereby enhances their antitumor activity. Another opportunity for improving T cell functionality came from the observation that SOCS1 KO enhanced CD4+ memory T cell proliferation. CD4+ T cell expansion enhanced CD8+ T cell cytotoxicity, and KO of SOCS1 dramatically improved the antitumor effect. The observed upregulation of Fas Ligand and TRAIL/TNFSF10 following SOCS1 KO led to the hypothesis that persistence of allogeneic T cells would be further improved by knocking out Fas and SOCS1, which was then demonstrated in the mouse model. Finally, Menger showed that SOCS1 KO also led to the upregulation of CISH and other SOCS family members. To inhibit the entire SOCS family, Menger and her team knocked out ZNF217, and found that this led to upregulation of the RNA m6A writer METTL3, which destabilized all of SOCS1, SOCS3, and CISH transcripts. ZNF217 KO in CD19 CAR T cells enhanced markers of stemness (IL-7R, CD27, and CD62L) and functionality (GZMB), and induced a gene signature similar to a recently observed signature associated with long-term response to CD19 CAR T cell therapy. In vivo, ZNF217 KO enhanced survival in mice bearing A549 lung adenocarcinoma tumors. A “molecular glue”, which enhanced ubiquitinoylation and subsequent degradation of ZNF217, provided a transient method to inactivate ZNF217, with multiple properties similar to the SOCS1 gene knockout in human T cells.
A TIGIT-CD28 chimeric switch receptor enhances TCR T cell function and metabolic fitness - Alexandra Reinbrecht, HI-TRON Mainz, Germany
T cell exhaustion is a major cause for failure of cellular immunotherapy. Alexandra Reinbrecht of Matthias Theobald’s lab set out to address this problem. She developed a chimeric switch receptor (CSR) consisting of the extracellular domain of TIGIT fused to the transmembrane and intracellular domains of CD28, thereby activating cells upon binding of CD155 on tumor cells to the extracellular TIGIT domain. The TIGIT/CD28 CSR enhanced tumor lysis of TCR-engineered T cells in repetitive coculture assays via an increase in mitochondrial functionality (which was observed across multiple donors) and cytokine responses. In vivo, the TIGIT/CD28 CSR significantly improved the efficacy of TCR T cells. To further evaluate the functionality of the TCR CSR T cells, Reinbrecht developed two 3D bioprint models by encapsulating the T cells with either tumor cells or tumor spheroids in an extracellular matrix. TCR CSR T cells demonstrated superior antitumor activity in these 3D models. Further testing will include different cell types to mimic the tumor microenvironment, and TCR CSR T cells derived from patient cells.
Antigen discovery and cancer antigen vaccination
Dissecting T cell recognition of human cancer - Ton Schumacher, Netherlands Cancer Institute, The Netherlands
Turning to understanding the fundamentals of T cell recognition of peptide:HLA as the “Grand Challenge” of cancer immunotherapy, Ton Schumacher described a highly scalable approach to ascertain the rules of this critical interaction, and the most relevant targets – the mechanistic basis of immune checkpoint therapy. To highlight the importance of neoantigens derived from personal mutations to cancer immunotherapy, Schumacher described the neoantigen responses in a patient with a complete response to tumor infiltrating lymphocyte (TIL) therapy. Mutation analysis, epitope predictions, and multimer screening identified a number of neoantigen responses of varying abundance (including up to 30% of all T cells in the TIL product being specific for one single epitope), which persisted for over a year. A key challenge going forward for the field though is to readily identify what specific neoantigens the patient does or can respond to as a biomarker and as guide for therapy, such as for personal cancer vaccines or adoptive cell therapy. Overcoming the vast combinatorial complexity is a significant challenge and has been first approached by sequence-based and structure-based approaches using data on epitope:TCR pairs found in the VDJdb database, with overall disappointing results. Schumacher pointed to the heterogeneous and highly non-systematic data as a possible cause for that poor performance, and so tested the reported epitope specificities using a high-throughput TCR screening approach they developed and validated. For nearly half of the TCRs reported in the VDJdb database (representing the 10 most densely covered epitopes), only 25-75% of the TCRs were reactive to the corresponding epitope, dramatically reducing the potential accuracy of any algorithm developed with the expectation these were valid TCR-epitope pairs. AlphaFold3 predictions of TCR::peptide:MHC structures showed significantly higher structural confidence for validating compared to non-validating pairs. Modeling the validating and non-validating pairs and looking for confidence of structural prediction between the TCR and peptide:MHC allowed development of the algorithm TCRBridge to predict effective pairing, which performed well (based on ROC curves). The specificity of TCRBridge was further validated by switching epitopes binding to the same HLA. Wrapping up, using a set of validated and non-validated TCR:epitope pairs identified with PairScan – a method to test for reactivity between libraries of epitopes and TCRs (200 x 200 in this case) – TCRBridge showed good separation between the validated and non-validated pairs. Overall, this scalable, semi-synthetic approach holds great promise to begin unraveling the TRC::peptide:MHC code based only on genetic sequence information.
The long and winding road towards personalized T cell therapy of pancreatic cancer - Rienk Offringa, German Cancer Research Center, Germany
Targeting pancreatic cancer is challenging due to its fibrotic, immunosuppressive tumor microenvironment, making primary tumor eradication difficult without surgery. Rienk Offringa focused on surgically resectable disease, which still has high recurrence rates. Analysis of large sections overcame a technical problem of poor detection of T cells in the abundant fibrotic areas. Offringa and his team found abundant T cells in two-thirds of pancreatic cancer tissues, often near tumor cells. These T cells, though exhausted, could be isolated and expanded, and showed in vitro responses to autologous tumor cells. Furthermore, dense tertiary lymphoid structures (TLSs) could be observed in approximately half of the tumor samples, correlating with better prognosis post-resection. TCR deep sequencing revealed expanded tumor-infiltrating lymphocyte (TIL) repertoires distinct from blood. However, isolating functional alpha-beta TCR pairs and maintaining dominant tumor-reactive clones during expansion was challenging, the latter due to T cell exhaustion. Offringa prioritized agnostic TCR immunity testing against autologous tumor cells, rather than predicted neoepitopes. Single-cell sequencing of TILs revealed an exhausted cluster of highly expanded clonotypes. These exhaustion gene signatures predicted tumor-reactive clonotypes with high accuracy (and conversely clonotypes with a bystander signature were not tumor-reactive), leading to the identification of tumor-reactive TCRs in both MSS and MSI pancreatic and colorectal cancer samples. After testing for reactivity to predicted neoantigens failed, Offringa turned to mass spectrometry, immunopeptidomics, single-HLA knockout tumor lines, and expression screening to identify target antigens and HLA restrictions. TCRs recognizing HLA-restricted private and recurrent T cell epitopes were identified from 3 pancreatic cancer samples, including one that also mediated activity against lung and colorectal cancer cell lines. The cognate private epitope of an HLA-B51-restricted TCR was identified to be encoded by an inactivating private frameshift mutation in the CDKN2A tumor suppressor gene. For a recurrent HLA-A2 epitope, Offringa and team couldn't annotate any peptides, potentially indicating an epitope derived from the "dark proteome”. Analogous studies in a clinically relevant mouse tumor model identified tumor-reactive TCRs, some against recurrent epitopes, and others private. One of the recurrent epitopes was derived from an endogenous retrovirus, which was only found in the particular transplant tumor model the researchers used. Based on the lack of actionable recurrent epitopes identified so far, they shifted to an autochthonous model that mimics human pancreatic cancer histology and allows for genetic manipulation and testing of (neo)adjuvant therapies. BRCA or mismatch repair deficiencies yielded tumors with an increased mutational load, as expected, but most of the identified TCRs were pan-reactive. In retrospect, this may have been caused by the low allele frequency of spontaneously-induced mutations, which only arose while the tumor was growing. cDNA expression screening ultimately identified a true tumor-specific self-antigen encoded by an orphan G-protein coupled receptor. While challenging to identify, the advantage of Offringa’s approach is that these TCR-targeted antigens are actually presented in the tumor, increasing their clinical relevance.
Challenges and advances in neoantigen discovery - Michal Bassani-Sternberg, Ludwig Institute for Cancer Research, Switzerland
Michal Bassani-Sternberg uses omics approaches to identify immunogenic antigens for cancer clinical trials, facing challenges in predicting responses and rapidly manufacturing personalized therapy. Her proteogenomics approach uses tumor immunopeptidomics and next-generation sequencing of tumor and blood to discover neoantigens. An end-to-end pipeline in a single container that integrates various different steps was developed. From single, multiple, or longitudinal biopsies, “NeoDisc” processes sequencing and mass spec data; performs alignments, mutation calling, HLA-typing, and transcriptome analysis; generates a personalized reference database; detects defects in the antigen presentation machinery; and identifies and predicts neoantigens for class I and II presentation. Further, NeoDisc designs optimal long peptides for vaccine manufacturing, producing an informative report, all within two weeks. This publically available pipeline identifies tumor-presented peptides from various sources, including tumor-associated antigens and tumor-specific antigens derived from mutations, oncogenic viruses, or non-canonical elements. The initial rule-based method for ranking immunogenic antigens, which considered binding affinity, presentation, and gene expression, became overly complicated as more parameters were added. Consequently, a machine learning algorithm was developed to identify important features and rank antigens based on their immunogenicity. Bassani-Sternberg and her team used a large, unbiased training dataset from Paul Robbins' group, which had called and experimentally confirmed mutations for 120 patient tumors. The algorithm was further trained and tested on additional internal and independent datasets, including a dataset from an HPV-positive cervical adenocarcinoma patient with nearly 400 called mutations. Benchmarking against the previous rule-based approach and other available pipelines showed superior performance, with the machine learning algorithm pushing verifiable immunogenic mutations higher up in the ranked list. Concurrently, an approach (NeoScreen) was developed to differentially enhance expansion of neoantigen-specific T cells by adding autologous engineered B cells as antigen-presenting cells (APCs), expressing costimulatory molecules and loaded with neoantigenic peptides (the top 50-100) from tumor fragment cultures. To illustrate antigen selection challenges, Bassani-Sternberg presented patient cases from the neoantigen-enriched TIL-ACT phase I clinical trial, NeoTIL, run in collaboration with George Coukos. A melanoma patient achieved a complete, durable response, but the target of the TCR that dominated the infusion product and persisted in the patient did not recognize any of the stimulating neoantigen peptides and remains unknown. For a lung cancer patient with a mixed response, therapeutic resistance was driven by tumor clones losing the dominant neoantigen target. A patient with stable melanoma eventually progressed due to B2M loss of heterozygosity and HLA-I expression, a known immune evasion mechanism. While mass spectrometry identifies tumor-presented antigens, they are not always highly ranked by the algorithms. To address this, a revised workflow (NeoDiscMS) involves sequencing first to get a list of predicted neoantigens, then the mass spectrometer performs a quick scouting scan of HPLC fractions and a real-time database search against the target list. Only if this scan passes certain cutoffs does it trigger the time-consuming, highly sensitive fragmentation scan for accurate identification of these antigens, while still generating discovery scans for other peptides. This approach significantly increases the number of data points, identification scores, and fragment coverage for targeted neoantigens, and improves the confidence for vaccine inclusion.
RNA vaccines for pancreatic cancer - Vinod Balachandran, Memorial Sloan Kettering Cancer Center, USA
While pancreatic ductal adenocarcinoma (PDAC) is an immune-cold tumor type that is not typically considered to be suited for vaccination due to low numbers of mutations and neoantigens, recent work has shown that in rare PDAC survivors, neoantigens arising from passenger mutations were targeted by endogenous CD8+ T cells, with evidence supporting a role for tertiary lymphoid structures (TLSs) in inducing such cells. Based on these findings, Vinod Balachandran and colleagues developed a novel mRNA vaccine targeting passenger mutation-derived neoantigens. In a clinical trial, personalized mRNA neoantigen vaccines containing up to 20 HLA class I/II neoantigens were administered following complete surgical resection of tumors. Patients were also treated with a single dose of anti-PD-L1 one month prior to vaccination, and with chemotherapy between the 8 priming doses and one booster dose 30 weeks later. T cell responses to the individual neoantigens in each patient were measured by ex vivo IFNγ ELISPOT, and confirmed using CloneTrack (a tool quantifying TCR expansion based on Vβ sequencing). High-magnitude T cell responses (up to 10% of total blood T cells) were identified in 50% of treated patients. Detailed analysis revealed that all responding patients had expanded clones that were vaccine-specific and primarily CD8+. Indeed, in one patient, a single vaccine-specific clone was undetectable prior to vaccination, and represented 1% of all T cells after vaccination. Further, positive immunological responses (defined as both ELISPOT positivity and clonal expansion) were correlated with lack of tumor recurrence. Interestingly, vaccine (and clinical) non-responders were enriched in patients who had undergone splenomegaly, suggesting an important role of the spleen with this i.v. delivered vaccine. To better understand the origin of the T cell responses, and whether they persist long-term, Balachandran and colleagues have evaluated peripheral blood, tumors, tdLNs, and tissue samples taken starting prior to and for nearly 4 years after treatment, allowing for tracking of all 79 vaccine-induced T cell clones across the 8 vaccine responders. Only 2 were detectable in the blood prior to vaccination, and clonal dynamics indicated that clones emerged with a median lifespan of 1 year after priming, and of nearly a decade after boosting, despite intervening chemotherapy. Detailed deconvolution of expanded clones has so far identified the targeted neoantigen for 20% of the expanded clonotypes. While avidity of mutated epitopes was generally higher than for the corresponding wild-type epitope, longevity of clonotypes was higher for those clonotypes with avidity closer to the wild-type epitope. Vaccine-induced clonotypes were almost exclusively induced de novo following the vaccine, while clonotypes induced by the anti-PD-L1 treatment were largely pre-existing. Single-cell RNA and TCR sequencing of most of the expanded clonotypes demonstrated that cells had a proliferative phenotype shortly after vaccination, rapidly contracted to an effector phenotype, and then transitioned over a longer period of time to a tissue-resident phenotype. Functionally, cells isolated up to 4 years after vaccination retained polyfunctionality and cytotoxicity for 6 of the 8 responding patients, which was observed for both the dominant and the subdominant clonotypes. Two patients who generated strong vaccine responses and did not recur in the 1.5-year follow-up did recur during the extended follow-up of 3 years. In these two patients, the loss of function appeared to be associated with a loss of specific T cells in the peripheral blood. Phylogenetic mapping of mutations in these two recurring patients indicated loss of vaccine-targeted neoantigen mutations, providing evidence for on-target vaccine-induced immune activity. A global randomized phase 2 clinical trial is in progress.
Neoadjuvant immunotherapy
Neoadjuvant immunotherapy in lung cancer: preclinical models, clinical trials and reverse translation - Tina Cascone, MD Anderson Cancer Center, USA
Tina Cascone began her talk by outlining three compelling rationales for neoadjuvant immune checkpoint blockade (ICB): (1) opportunity for robust up-front treatment, potentially combined with chemotherapy to maximize the effect of tumor cell death and activation of a diverse T cell response, (2) readout of impact on the surgical specimen, and (3) potential for translational analyses. These benefits may be extended with perioperative ICB by countering some of the immunosuppressive effects of surgery and further enhancing antitumor immunity. Early preclinical work had shown that survival following neoadjuvant ICB was superior to adjuvant ICB – marked by enhanced T cell responses – and identified PD-L1 as a facilitator of metastases in murine lung cancer models. Stressing that patients with resectable non-small cell lung cancer (NSCLC) are at high risk for disease recurrence within 12 to 18 months post-surgery, Cascone extended this earlier work into the highly metastatic NSCLC KP-OVA model and again demonstrated the benefit of neoadjuvant versus adjuvant ICB treatment, including a reduction in metastatic disease burden. The NEOSTAR randomized phase 2 clinical trial evaluated neoadjuvant treatment with anti-PD-1 vs anti-PD-1/anti-CTLA-4 combination in patients with untreated, resectable NSCLC, and showed that the combination treatment met the pre-specified endpoint, with 8/21 patients achieving major pathological responses (MPR; ≤ 10% residual tumor), 6 of whom had pathological complete responses (pCR). These results were extended in a platform trial (no randomization) comparing single or combination ICB with doublet chemotherapy (CT) in lung cancer, which again demonstrated superiority of the combination arm. Correlative analysis, an important part of the strategy Cascone developed to inform future trials, demonstrated improved tumor infiltration with T cells, including CD8+ effector memory and tissue-resident memory T cells in post-surgical samples following combination treatment. These improvements were confirmed by single-cell RNA and TCR sequencing for a subset of patients. T cell, B cell, and tertiary lymphoid structure (TLS) scores were increased in resected tumors treated with the ICB combination + CT, and from responders compared to non-responders. Focusing on the role of TLS in neoadjuvant therapy, Cascone showed that B cells, plasma cells, and TFH cells increased in the responders. Moving forward, the NeoCOAST trial was designed to test other novel immunomodulatory agents (anti-CD73 antibody, anti-NKG2A antibody, and anti-STAT3 ASO) that had shown promise in unresectable disease as neoadjuvant therapies in resectable disease in combination with anti-PD-L1 treatment. Again, the MPR rates of the combination arms were significantly higher (22% - 33%) compared to the anti-PD-L1 arm alone (12.5%). Single-cell sequencing of the antibody combination arms showed increases in CD8+ T cells, NK cells, cytotoxic markers, TLSs, and the T cell recruitment marker CXCL13. NeoCOAST-2 was then designed to evaluate the anti-CD73 and anti-NKG2A antibodies on top of the now standard-of-care neoadjuvant anti-PD-(L)1 + CT and adjuvant anti-PD-1 treatment in a randomized design. Importantly, this study also included a new arm with neoadjuvant Dato-DXdm, an antibody–drug conjugate (ADC) targeting Trop2, plus anti-PD-L1 and CT, and adjuvant anti-PD-L1. The ADC demonstrated the greatest increases of both MPR and pCR, potentially due to increases in immunogenic cell death. Finally, a multi-institutional collaboration is underway to study how TLSs develop and work in the neoadjuvant ICB setting. For this, Cascone and her team have established a novel human-relevant orthotopic NSCLC murine model to examine TLSs, germinal centers, and B cell–T cell interactions within the context of both T and B cell neoantigens. Furthermore, multi-omics analyses are currently being conducted to translate findings between the lab and clinic.
Overcoming immunotherapy resistance
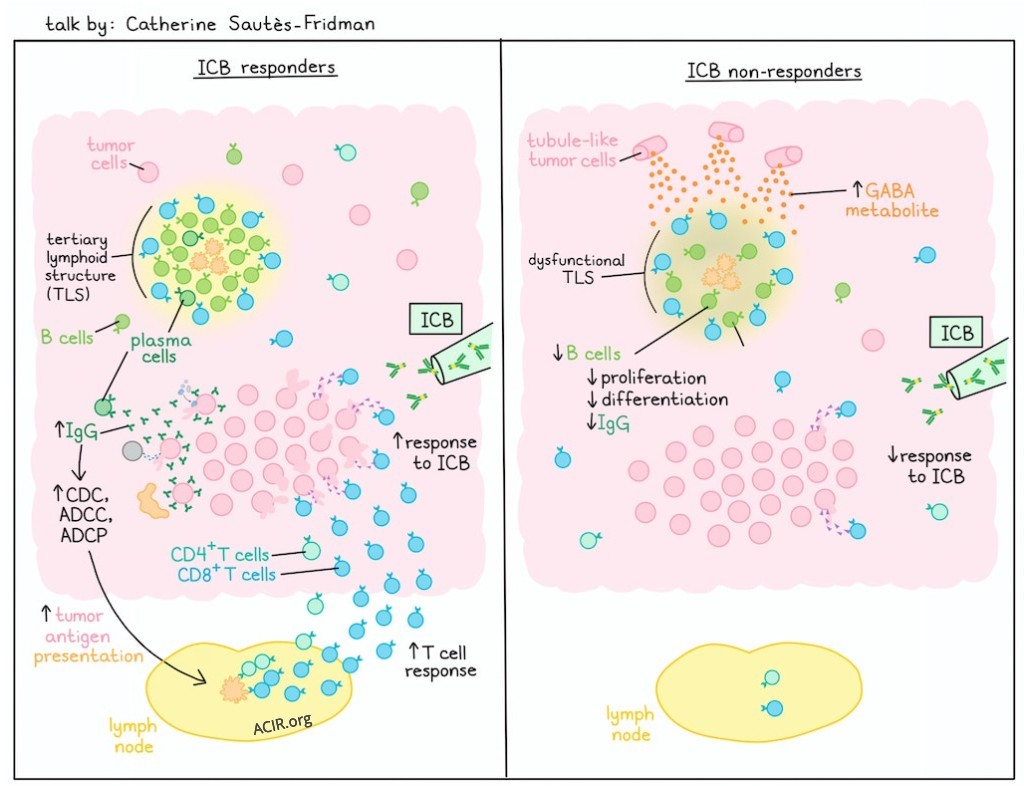
Spatial determinants of tumor immunity and immunotherapy resistance in renal cell carcinoma - Catherine Sautès-Fridman, Cordeliers Research Centre, France
Uncovering a novel mechanism of immune resistance, Catherine Sautès-Fridman described how the derivatives of the neurometabolite gamma amino butyric acid (GABA) impact the immune system. She began by describing their studies on the role of tertiary lymphoid structures (TLSs) using samples from a cohort of patients with metastatic renal cell carcinoma (RCC) who were treated with immune checkpoint blockade (ICB). Mature TLSs in tumors have been shown to be key intratumoral structures associated with positive responses to immunotherapy. Plasma cells could be detected in the TLSs and in the tumor beds, and histological staining demonstrated the presence of IgG-covered tumor cells, which was correlated with responsiveness to ICB. The antibodies decorating the tumor cells are proposed to stimulate ADCC, ADCP and/or CDC, leading to release of tumor cell antigens and immune complexes, initiating adaptive T cell immunity. Supporting this hypothesis, a clinical trial in ICB-treated patients with soft tissue sarcoma demonstrated the predictive value of selecting TLS+ patients, but Sautès-Fridman questioned why some TLS+ patients did not respond. She returned to the RCC cohort to conduct a detailed omics and preclinical analysis of ICB-responsive and non-responsive tumors. Bulk RNA sequencing demonstrated upregulation of B cell, Th2 cell, and macrophage activation, and IFN response in the tumors of responding patients, but interestingly, non-responders upregulated gene signatures of multiple GABA pathways (secretion, synthesis, transport). GABA is a neurotransmitter produced as a metabolite that can fuel the TCA cycle. A high GABA signature in this cohort of patients was associated with poor response to ICB and reduced progression-free survival (PFS). Similar results were observed in 3 other ICB-treated RCC cohorts. Moreover, the GABA-high signature was associated with a downregulation of MHC–TCR interactions, reduced type I IFN production and responses, a decrease in intratumoral and intra-TLS B cells, and low germinal center (GC) B cell proliferation. Analysis of single-nuclei RNAseq data showed that proximal tubule-like tumor cells were the dominant cell type bearing the GABA signature, and also demonstrated strong presence of the GABA metabolite. Nonetheless, the neuronal signature in Schwann cells and sensory neurons between responders and non-responders did not differ, indicating it was not a generalized effect on local neuron presence. Mechanistically, Visium sequencing analysis of TLS regions showed that non-responders exhibited decreased IgG and MHC I/II, and increased IgA expression, suggesting dysfunction in immune activity. Spatial analysis demonstrated increased adjacency of dysfunctional TLSs to GABA-producing proximal tubule-like tumor cells. Digging deeper, TLSs near to proximal tubule-like tumor cells showed gene signatures associated with defective antigen presentation, T cell activation, B cell differentiation, and accumulation of cycling GC B cells, and an upregulated TCA cycle. The association with a high GABA signature and poor response was also demonstrated in the soft tissue sarcoma cohort. These findings were supported by studies in the MCA-OVA murine model which showed an improved response to immunotherapy and increased B cell infiltration into TLS in mice treated with 3-MPA, which inhibits GABA production. Wrapping up the mechanistic studies, exposure of human B cells to GABA in vitro led to reduced antibody production and proliferation.
Fc-optimized CD40 agnostic antibody for the treatment of cancer - Juan Osorio, Rockefeller University, USA
Although the critical role of dendritic cells in both the tumor and the lymphatic compartments is well appreciated, efforts to agonize CD40, one of the key DC activators, have so far struggled clinically due to both low stimulatory activity and systemic toxicity. Agonizing CD40 requires assembling a CD40-trimer, an intrinsic effect of the trimeric CD40 ligand, but not readily accomplished with bivalent anti-CD40 antibodies. Juan Osorio described their efforts using Fc-engineered anti-CD40 antibodies designed to more effectively bind the FcγR on immune cells to efficiently multimerize and cross-link CD40 on dendritic cells, leading to enhanced DC and T cell activation. A combination of five Fc mutations in the parental 2141 antibody led to a construct (2141-v11) with ~100-fold enhanced selective binding to FcγRIIB and >10-fold enhancement in antigen responses, but which still induced thrombocytopenia when delivered systemically. To overcome this limitation, Osorio turned to intratumoral (i.t.) delivery in a first-in-human, dose-escalation clinical trial in patients with measurable, advanced metastatic disease with accessible lesions. 2141-v11 was tolerable, with no dose-limiting toxicities (even at the maximal dose tested) and no appearance of thrombocytopenia or liver toxicity. Even in this heavily pre-treated population, half the patients (6/11) experienced a reduction in tumor size, with two patients demonstrating a complete response. Responses continued for a median of 12 months, with one patient remaining free of cancer for more than 2 years. In a responding patient with breast cancer, beyond elimination of disease in the injected skin lesion, tumors in the lymph node and the liver were eradicated, and multiple systemic markers (e.g., CA-125) were dramatically and stably reduced. There was no difference in the baseline number of CD8+ T cells in the two complete responders versus the non-responders, but following treatment, increases in total, effector, effector memory, and granzyme B+ CD8+ T cells were observed, as well as a dramatic increase in significantly expanded CD8+ clonotypes. Post-treatment biopsies of responders also demonstrated increases in mature tertiary lymphoid structures (TLSs) containing CD8+ and CD4+ T cells, CD11c+ DCs, and B cells. In a CD40- and FcγR-humanized, bilateral murine tumor model, 2141-v11 led to rejection of both injected and non-injected tumors, and protective memory. Mechanistically, i.t. injection of 2141-V11 led to TLSs in injected, but not non-injected, lesions, which contained CCR7+ DCs. Furthermore, injected tumors showed an increase in effector CD8+ T cells, which were shared between injected and non-injected lesions. Based on FTY720 treatment, tumor rejection in injected tumors was independent of lymphoid tissue-derived T cells, but non-injected tumors depended on lymphoid tissue-derived cells. In a recent phase 1/2 trial of intravesicular 2141-V11 for BCG-resistant non-muscle invasive bladder cancer, 38% of patients experienced a durable complete response at 6 months (one ongoing for 4+ years) and almost all (92%) patients showed absence of progression. Finally, analysis of TLSs in these patients shows variability, which appears related to response.
Cytotoxic PD-L1/PD-L2 dual-specific antibodies couple tumor stroma remodeling with checkpoint blockade to drive rejection of “cold” cancers - Michael Curran, UT MD Anderson Cancer Center, USA
Current oncology efforts, driven by PD-1 blockade, show lower response rates in many cancers (e.g., pancreatic, ovarian, prostate) than those observed in melanoma. To improve outcomes in "cold" tumors, understanding and overcoming barriers is crucial. While PD-1 and PD-L1 are targeted, PD-L2 has been historically underappreciated, partly due to biological differences between mice and humans. In humans, it's widely expressed on stroma, tumor, and endothelial cells in various cancers, and binds to PD-1 with a 5x higher affinity than PD-L1, contributing to checkpoint efficacy. Recognizing that PD-L1 and PD-L2 harbor a conserved structure involved in PD-1 engagement, and the potential importance of blocking PD-L2 interactions, Michael Curran set out to develop a single antibody that would target both PD-L1 and PD-L2 to silence the entire inhibitory circuit – interactions between PD-L1 and PD-1, PD-L2 and PD-1, and B7.1 and PD-L1. Two initial hits were identified, and while initial affinities were low, maturation steps increased the potency of both to subnanomolar levels for both targets. The fully human dual-specific PD-1/PD-L2 antibodies (DiPDL) demonstrated superior blockade of T cell inhibition compared to commercial anti-PD-L1 and anti-PD-1 antibodies, and only DiPDL3 also blocked PD-L1/B7-1 cis-interaction. Thus, DiPDL3 became the starting point for the clinical candidate IMGS-001. The goal was to transform suppressive tumor microenvironments into more permissive settings by activating immune cells, including NK cells and neutrophils. Since a big barrier to checkpoint efficacy in cold tumors is immune exclusion, the Fc was modified to induce ADCC and ADCP activity. In immunoincompetent xenograft models, DiPDL was able to significantly slow tumor growth solely based on cytotoxic activity. In vivo studies in anti-PD-1-refractory B16 melanoma showed tumor regression and cure rates between 30 and 70%, significantly outperforming existing anti-PD-1 antibodies. This efficacy was attributed to the reduction of immunosuppressive myeloid cells and an increase in CD11c+ DCs and CD8+ T cells in tumor-draining lymph nodes and tumors. Depletion of M2 macrophages, potential remodeling of the PD-L1/PD-L2+ tumor vasculature, and killing of PD-L2+ cancer-associated fibroblasts “opens” the TME and enables the infiltration of T cells into the tumor core. Even in the "hot" CT26 tumor, where PD-1 blockade is effective, DiPDL offered a significant additional advantage through its effect on multiple immunosuppressive cells. Preliminary clinical data from a phase 1a/1b trial showed promising results in two patients who had failed on prior anti-PD-1 therapy. The first 15 patients experienced no greater than grade 1 immune-related adverse events, and grade 2 or higher infusion-related reactions were manageable.
In situ cancer vaccination
Enabling in situ vaccination with TL7/8/9 RNA/DNA agonists - Arthur Krieg University of Massachusetts, RNA Therapeutics Institute/Zola Therapeutics, USA
Coley’s toxin and BCG vaccines activate antitumor immune responses through TLR9-mediated detection of bacterial DNA , specifically unmethylated CpG residues, which are typically highly methylated in vertebrates. However, early cancer trials using synthetic CpG TLR9 agonists (CpG-B or -C) failed, leading Arthur Krieg to develop CpG-A (vidutolimod). CpG-A features a phospodiester backbone susceptible to what is now known to be necessary DNAase II cleavage (compared to the DNAase-resistant phosphorothioate in CpG-B/C). It also contains poly-G regions that form G-quadruplex structures, which induce a stronger IFNα response. Intratumoral monotherapy with CpG-A packaged in virus-like particles (VLPs), showed promising results in advanced PD-1-refractory melanoma, including regressions in distant sites, and as neoadjuvant therapy. However, neutralizing antibodies against the VLPs were observed, which inhibited IFNα induction. To overcome this, lipid nanoparticles (LNPs) were subsequently used as a non-immunogenic delivery vehicle. In addition, translational data revealed high numbers of tumor-associated macrophages (TAMs) as a predictor of resistance to vidutolimod. Krieg described how immune cells differentiate viral particles from dying cell debris by detecting GU-rich RNA (via TLR7/8) and unmethylated CpG dsDNA (via TLR9), which are uniquely found in viral particles compared to damaged cell debris. Various immune cells then cooperate and eventually induce a CD8+ T cell response, which is crucial for clearing viral infections – and for efficient cancer immunotherapy. Hypothesizing that TLR 7/8/9 evolved as a protective mechanism against genome damage, Krieg developed Z-007 as a “synthetic retrovirus” comprised of CpG-A and GU-rich RNA packaged in an LNP, hoping to mimic a viral infection and reprogram TAMs and the TME to induce CD8+ T cells. In human tumor-associated ascites cells, Z-007 induced the production of very high levels of IFNα, with low levels of inflammatory cytokines (IL-1β, IL-10, TNFα), decoupling the classic inflammatory responses. Pharmacodynamic studies of intravenous (i.v.) Z-007 in mice and cynomolgus monkeys revealed induction of IFNα (at high levels), PD-L1, and M1 macrophage and IFNγ transcriptional signatures. No signs of toxicity were observed. Treatment of companion dogs who had spontaneously developed cancer with i.t. or i.v. Z-007 was safe, and led to significant tumor reduction, even in tumors that were resistant to chemotherapy and prior treatment with TLR agonists. Krieg is now focused on the human clinical development of Z-007 for systemic administration. Human pharmacokinetics indicate that i.v.-administered LNPs predominantly go to the liver, which is an immunosuppressive environment. Z-007 may be able to reprogram liver TAMs, induce a response to liver metastases, and promote systemic tumor regression.
By Ute Burkhardt, Ed Fritsch, and Lauren Hitchings



