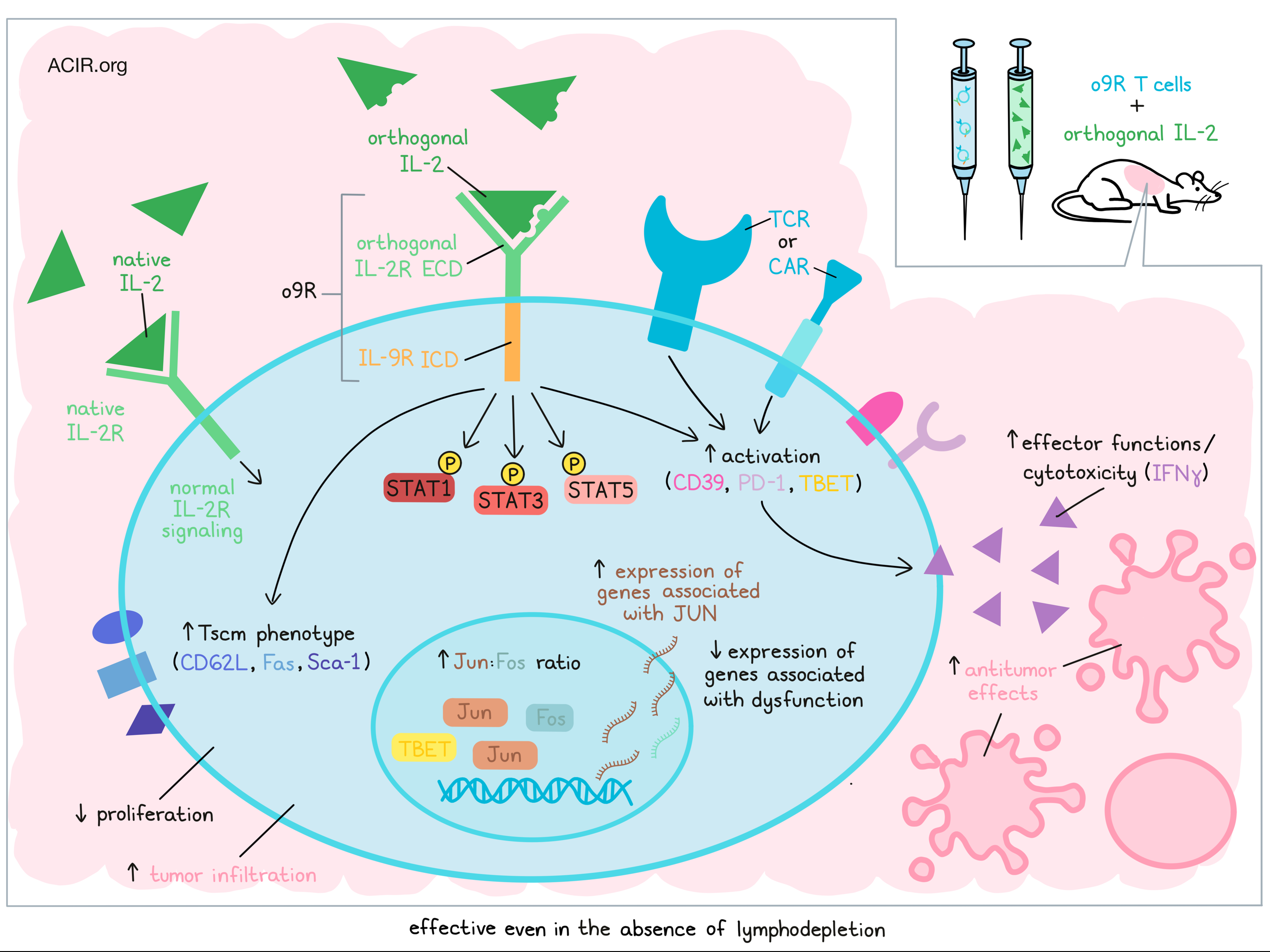
Adoptive T cell therapy has come a long way in recent years, but it typically requires lymphodepleting pre-treatment, which can limit patient eligibility, and its efficacy is still limited in many patients. In recent efforts to improve ACT, particularly for solid tumors, Kalbasi, Siurala, and Su et al. engineered T cells with orthogonal cytokine receptors – mutated versions of native cytokine receptors that selectively bind mutated versions of their corresponding cytokines. In this case, the engineered receptors were chimeric, containing an extracellular domain of orthogonal IL- 2R (oIL-2R) (which binds only to an orthogonal version of IL-2), and various intracellular domains from receptors for IL-2, IL-4, IL-7, IL-9, or IL-21. This allowed the researchers to use previously developed oIL-2R/oIL-2 cytokine pairs to induce and study the effects of different γc cytokine receptors in adoptively transferred engineered T cells, without inducing systemic cytokine signaling. Their findings were recently published in Nature.
Stimulation of chimeric orthogonal receptors with oIL-2 fused to mouse serum albumin (MSA-IL-2) induced patterns of phospho-STAT signaling that were consistent with known signaling for each γc cytokine receptor, without interfering with wild-type IL-2-induced STAT signaling. Cells expressing the receptor with the intracellular domain for IL-9R (o9R) showed increased STAT1, STAT3, and STAT5 phosphorylation, consistent with natural IL-9R signaling, which has only a limited role in T cell biology. These cells also showed increased expression of CD62L, Fas, and Sca-1, consistent with a stem cell memory (Tscm) phenotype, and reduced proliferation compared to control o2R T cells (containing the IL-2R intracellular domain).
Looking more closely at o9R signaling, the researchers incorporated the o9R receptors into pmel (gp100-specific) T cells and adoptively transferred them into B16 melanoma-bearing mice without lymphodepleting pre-treatment. In this setting, both o9R and o2R cells expanded and showed proliferation and STAT signaling profiles that were consistent with in vitro observations. Compared to o2R cells, o9R cells treated with MSA-oIL-2 induced more consistent antitumor effects and prolonged survival comparable to the effects of pmel T cells with lymphodepletion. o9R cells treated with MSA-oIL-2 were also superior within the context of lymphodepletion.
Given that oIL-2 is highly orthogonal and does not interact with endogenous cells, the researchers hypothesized that dosing with oIL-2 would be more tolerable than with IL-2, which becomes toxic after about 5 days. Indeed, in a prolonged dosing regimen of 25 days, no clinical toxicity was observed, and o9R T cells without lymphodepletion induced better antitumor results than pmel ACT with lymphodepletion and 5 days of MSA-IL-2.
Investigating the underlying mechanisms behind the superior antitumor efficacy of o9R pmel T cells, despite their weaker capacity for proliferation, the researchers found that at five days after transfer, o9R cells showed greater tumor infiltration and made up a higher percentage of CD45+ TILs compared to o2R cells. They were also enriched for clusters associated with T cell activation, including a cluster that expressed CD39, PD-1, and TBET. In vitro, o9R showed a higher cytotoxic capacity and increased IFNγ production compared to o2R cells.
RNAseq analysis showed that compared to o2R pmel T cells, o9R cells were enriched for genes associated with a Tscm phenotype, as well as for genes associated with activation and effector functions, which are typically excluded from the Tscm phenotype. A transcriptomic analysis of transcription factor pathway enrichment showed significant enrichment for genes associated with STAT1 and STAT3, as expected, as well as genes associated with JUN. This was accompanied by an increase in the ratio of Jun to Fos expression, which indicates tumor-specific T cells that are resistant to tumor-mediated exhaustion. In line with this, genes associated with T cell dysfunction were downregulated. Whether these features represented a distinct T cell phenotype or heterogeneity was unclear.
Looking specifically at CD62L expression, the researcher saw that while expression levels were higher on o9R cells in draining lymph nodes and spleens, they were comparable on o9R and o2R cells in the tumor, suggesting that antigen-specific T cell activation may dominate in the TME.
Kalbasi, Siurala, and Su et al. next tested the effects of o9R in CAR T cells and found that CAR-o9R T cells resembled o9R pmel T cells, showing characteristic STAT phosphorylation, reduced proliferation, and increased stemness, activation, effector functions, and antitumor activity in vitro. Using lymphodepleted mice bearing immunotherapy-resistant pancreatic tumors expressing mesothelin, the researchers tested adoptive transfer of mesothelin-specific CAR T cells engineered with o9R. In contrast to previous experiments, in which MSA-oIL-2 was administered systemically, the researchers switched to adenovirus-vectored intratumoral delivery of oIL-2 (Ad-oIL-2) to ensure high concentrations of oIL-2 in the tumor. In this setting, CAR-o9R T cells plus Ad-oIL-2 induced early toxicity, with 5 of 12 mice dying by day 10. However, 6 of 12 mice achieved complete regressions, which appeared to be more durable than the complete regressions (8 out of 12 mice) induced by CAR-o2R cells plus Ad-oIL-2.
Evaluating the high toxicity, which was consistent with immune effector cell-associated neurotoxicity (ICANs), the researchers found evidence of on-target, off-tumor activity against mesothelin-expressing meningeal cells, as well as increased mesothelin expression, which can occur in the context of inflammation. Evidence of cytokine release syndrome and tumor lysis syndrome were not observed, suggesting that the toxicity was mostly related to the specificity of the CAR.
Interestingly, when the researchers tested the same treatment without lymphodepletion, the toxicity of CAR-o9R was reduced. In this setting, the CAR-o9R cells outperformed the CAR-o2R cells, inducing more complete regressions and prolonging survival. Compared to the pmel T cell experiments, tumor infiltration of transferred cells was less pronounced, possibly as a result of intratumoral, rather than systemic delivery of oIL-2. Still though, the frequency of intratumoral CAR-o9R cells expressing IFNγ in the tumor was increased, suggesting that enhanced potency, rather than enhanced infiltration, may play a more important role in the improved antitumor efficacy.
Finally, to evaluate the translational potential of this approach, the researchers engineered human o9R and o2R T cells specific for NY-ESO-1. When treated with human oIL-2, the human o9R and o2R cells demonstrated characteristics that were consistent with what was seen in mouse cells. Differences between the two cell types persisted even after four challenges with a NY-ESO-1+ cell line, with the ho9R cells retaining a greater frequency of CD45RA+CD27+ and Tscm cells, and expressing higher levels of CD62L and CXCR3. Upon stimulation, ho9R cells were more cytotoxic and polyfunctional. Similar results were also observed in mesothelin-specific CAR-ho9R and CAR-ho2R cells.
Overall, these results show the potential to enhance adoptive T cell therapies using orthogonal cytokine and cytokine receptors pairs, and particularly, the o9R receptor paired with oIL-2, which enhanced antitumor immunity in a variety of models. However, given the enhanced potency and the potential for on-target, off-tumor toxicity, it will be important to incorporate additional safety features into future systems.
Write-up and image by Lauren Hitchings



