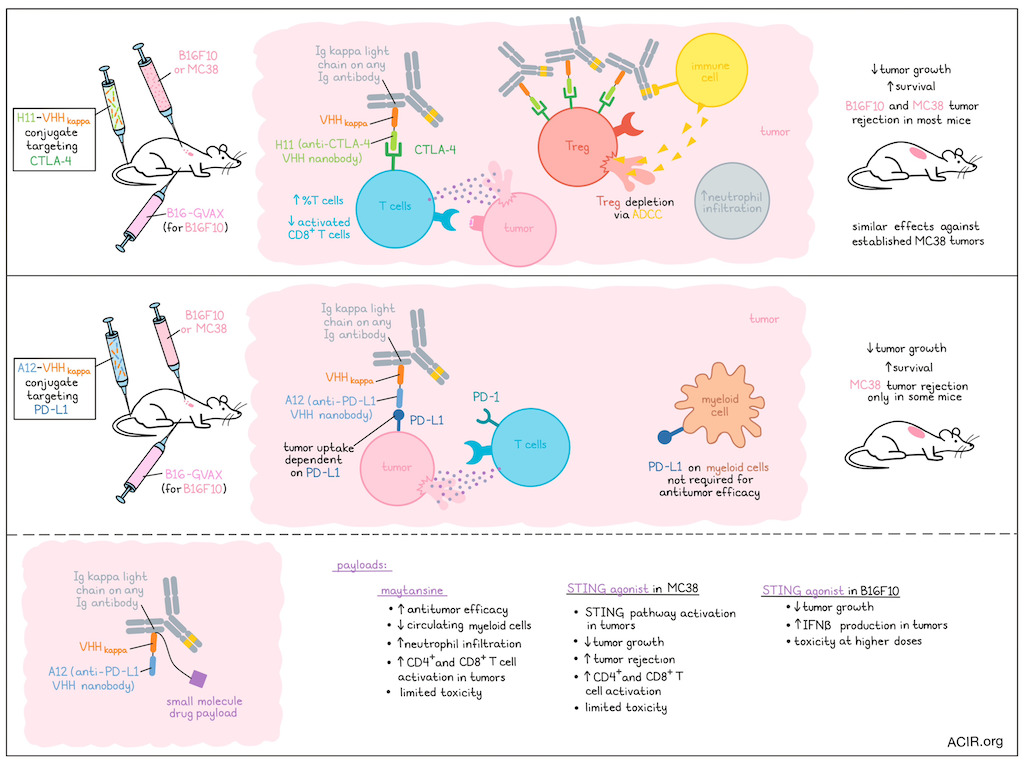
Various approaches have been taken or are underway to improve responses to CTLA-4- or PD-1/PD-L1-directed immune checkpoint blockade (ICB). Making use of nanobodies specific these checkpoints, a nanobody recognizing the murine Ig kappa constant chain (targeting the endogenous antibody repertoire), and nanobody–drug conjugate constructions, Liu, Le Gall, Alexander, et al. created various bispecific antibody engagers targeting CTLA-4 and PD-L1, and tested them for efficacy and safety in tumor models. Their findings were recently published in Nature Biomedical Engineering.
Nanobodies are the recombinantly expressed variable regions of heavy-chain-only antibodies (VHH). The researchers produced bivalent nanobody conjugates by genetic fusion of anti-CTLA-4 VHH (H11) or anti-PD-L1 VHH (A12) sequences to VHHkappa – a nanobody that recognizes Ig kappa light chains (used by >95% of all antibodies in adult mice) and can engage with any Ig isotype. Additionally, small-molecule drug payloads could be installed at the C-terminus of the conjugates. H11 and A12 maintained their affinity and specificity after conjugation, and the constructs could effectively recruit polyclonal Igs across different isotypes, as measured by surface plasmon resonance, and IgG, IgM, and IgA bound to B16F10 melanoma cells in the presence of the conjugates.
To assess antitumor activity, the anti-CTLA-4 VHH (H11)–VHHkappa conjugate was tested in the subcutaneous B16F10 melanoma and MC38 colon carcinoma models, with the conjugates injected intravenously beginning at the time of tumor cell injection. The melanoma model also received B16-GVAX cells at time of tumor inoculation, which produces GM-CSF and serves as a vaccine that enhances immunotherapy-induced antitumor responses, but does not slow tumor growth on its own. In both models, mice were treated with 3 weekly injections of the conjugates. In the B16F10 model, the H11–VHHkappa conjugate slowed tumor growth and improved survival, while a mixture of H11 and VHHkappa did not have efficacy. In the MC38 model, the conjugate was more effective than a commonly used anti-CTLA-4 monoclonal antibody (mAb 9H10). In the tumor microenvironment (TME), the H11–VHHkappa conjugate caused depletion of Tregs, while Tregs in the draining lymph nodes or spleen were not affected. The total percentage of T cells within the TME’s CD45+ population increased. When testing the conjugate in mice deficient in complement component C3 or lacking the FcR common γ chain, it was found that tumoral Tregs were depleted in the C3-deficient mice, as in the wild-type mice. However, this Treg depletion was impacted in the FcR-deficient mice, suggesting these effects were due to antibody-dependent cell-mediated cytotoxicity (ADCC). Compared to the anti-CTLA-4 mAb, the conjugate induced more neutrophil infiltration, which may play a role in the ADCC induced by the conjugate. Activated CD8+ T cells were reduced in both conjugate- and mAb-treated groups (due to their expression of CTLA-4).
The researchers then determined the antitumor activity of the conjugate on established MC38 tumors. Treatment was provided on days 7, 10, and 13 post-tumor injection. The conjugate again inhibited tumor growth and resulted in 100% survival.
Moving to testing of the anti-PD-L1 VHH (A12)–VHHkappa conjugate, imaging studies showed that the conjugate was taken up by MC38 tumors, and that uptake was PD-L1-dependent. This conjugate was also assessed for efficacy in the MC38 and B16F10 models beginning at the time of tumor cell injection. The conjugate slowed tumor growth and improved survival, while a mixture of A12 and VHHkappa did not have efficacy, and the conjugate was more effective than an anti-PD-L1 mAb in both tumor models. However, the conjugate only induced tumor rejection in some of the mice. Mice inoculated with tumor cells lacking PD-L1 did not respond to treatment, while mice that only lacked PD-L1 on myeloid cells responded to treatment in a similar way to WT MC38 tumors. Therefore, the expression of PD-L1 by myeloid cells was not required for efficacy of treatment.
To improve the antitumor effects of the anti-PD-L1 VHH (A12)–VHHkappa conjugate, an A12–VHHkappa drug adduct was generated for targeted delivery of the drug to the TME. The first payload tested was maytansine, a microtubule assembly inhibiting cytotoxic drug, which was attached via a disulfide cleavable linker. The maytansine-loaded A12–VHHkappa conjugate showed better antitumor activity than A12–VHHkappa in the MC38 model. No weight loss was detected, suggesting limited toxicity. The percentage of myeloid cells decreased in circulation, and in the tumor, there was an increase in neutrophil infiltration and CD4+ and CD8+ T cell activation. In the B16F10 model without the GVAX vaccine, this construct had antitumor activity and improved survival.
The researchers then created constructs using a cyclic dinucleotide STING agonist as the payload to help establish an inflammatory TME that aids in the antitumor response. In the STING agonist-loaded anti-PD-L1 VHH (A12)–VHHkappa conjugates effectively activated the STING pathway in the TME. In the MC38 model, treatment with the A12–VHHkappa–STING agonist reduced tumor growth and cured half of the mice, which was more effective than combined A12–VHHkappa conjugate and free STING agonist treatment. Again, no weight loss was detected. Treatment increased the percentage of activated CD4+ and CD8+ T cells in the tumors. In the B16F10 model,The A12–VHHkappa–STING conjugate was more effective than the A12–VHHkappa conjugate, but did not improve survival. At a higher dose, there was a stronger reduction in tumor growth and improved survival, but that dose increased body weight loss. The STING agonFormaist adduct, but not the A12–VHHkappa conjugate, resulted in IFNβ production in the tumor, but not elsewhere in the body.
Finally, the researchers performed a systemic toxicity analysis in mice for all developed constructs and did not detect any acute liver or renal toxicity.
Together, these data suggest that systemically delivered checkpoint-targeting nanobodies conjugated with a nanobody broadly targeting endogenous antibodies can be used alone or loaded with payloads, and can traffic to the TME to improve antitumor responses akin to ICB. This opens up new avenues for research into various combinations that might improve responses to these therapies in cancer types that remain largely resistant.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first authors Xin Liu and Camille Le Gall have answered our questions.

What was the most surprising finding of this study for you?
XL & CLG: One of the most surprising aspects of this study was how robust our platform turned out to be. The nanobody adducts recruit endogenous immunoglobulins, repurpose them as checkpoint blocking antibodies, and achieve potent checkpoint inhibition via targeting either PD‑L1 or CTLA‑4. In addition, the adducts can recruit immunoglobulins of any isotypes, which diversifies Fc-effector mechanisms to maximize immune cell activation and deplete checkpoint-overexpressing immune-suppressive cells. This simple strategy rivals traditional monoclonal antibodies in efficacy, manufacturability, and stability. We were pleasantly surprised by how well the bispecific nanobody adducts performed in vivo. They outperformed the benchmark therapy in multiple cohorts. That level of robustness was an eye‑opening demonstration of the design elegance and potential for clinical translation.
What is the outlook?
XL & CLG: The nanobody adduct production is easily scalable and more cost-efficient than monoclonal antibodies. In addition, nanobodies typically sustain lyophilization. We think this property is of particular interest because lyophilized biologics do not require strict cold-chain management. PD‑L1 and CTLA‑4 blockade have been clinically validated in various cancer types. If clinically applicable, our strategy could offer a more affordable (and possibly more effective) alternative to immune checkpoint immunotherapies. This would be particularly impactful in low‑resource settings. We hope this study will lead to further research into human immune‑reconstituted models and pharmacokinetics to determine clinical potential.
What was the coolest thing you’ve learned (about) recently outside of work?
CLG: My PhD advisor would be thrilled to know that I’ve been getting more and more into bird watching. Since moving to New England for my postdoc just over two years ago, I have been discovering how much native bird species differ from those in Europe. It’s so fun to learn about them and spot them on hikes or around the city!



