While personalized neoantigen vaccines have great potential, T cell responses induced by these vaccines often lack strength. Blass, Keskin, et al. aimed to improve T cell antitumor immunity by optimizing vaccine adjuvants and combining the vaccine with ICB. The results of their phase 1 trial in patients with melanoma were recently published in Cell.
The researchers hypothesized that they could (1) improve CD8+ T cell responses to personalized neoantigen vaccines by extending antigen exposure using a formulation with Montanide, a mineral oil-based adjuvant that induces local inflammation; (2) improve T cell priming by co-administering ipilimumab subcutaneously at the vaccine site; and (3) reduce immune suppression in the tumor by systemic nivolumab treatment.
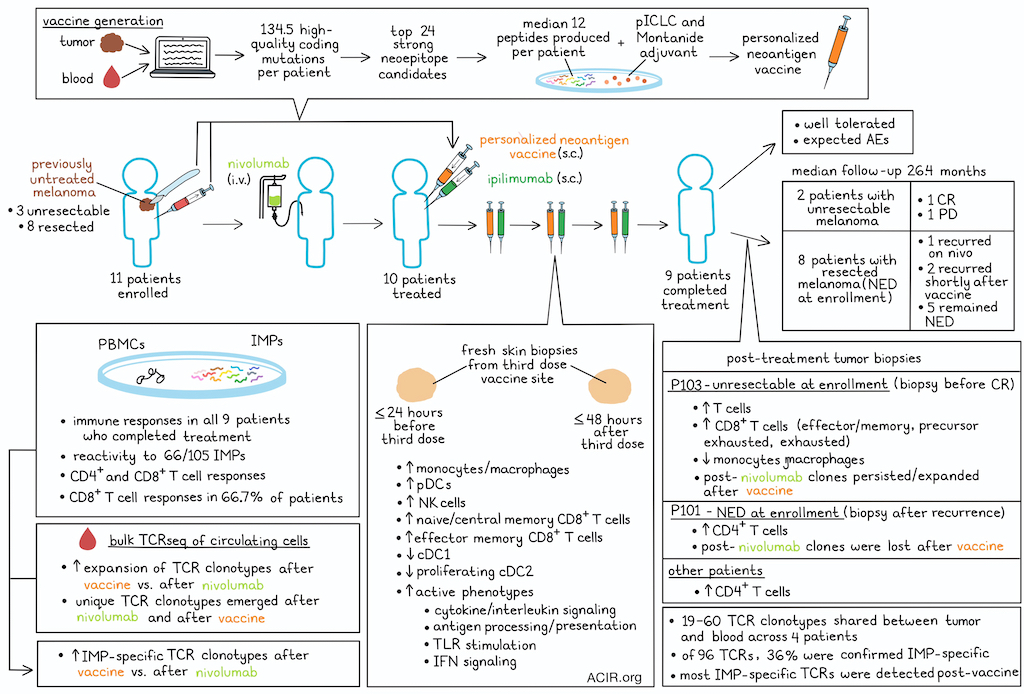
Eleven patients with previously untreated melanoma were enrolled, of whom 3 had unresectable and 8 had resected melanoma. Patients received nivolumab while the vaccine was being generated, which was successfully generated for 10 patients. A median of 134.5 high-quality coding mutations were detected per patient, and ranking based on a weighted heuristic considering binding prediction, gene expression, and cancer cell fraction led to selection of the top 24 strong neoepitope candidates, of which a median of 12 (range of 8–19) peptides 16–26 amino acids in length were successfully produced per patient and allocated to 2 peptide pools per patient.
Ten patients received the vaccine and ipilimumab (2.5 or 5 mg). Nine patients completed the full series of 4 treatments, while one patient dropped out due to rapid progression. Treatment was well tolerated, without unexpected adverse events. Of the two patients with unresectable melanoma, one had a complete response (CR), while the other patient had progressive disease. Of the eight patients who had no evidence of disease (NED) at the time of enrollment, 1 experienced recurrence on nivolumab alone, 2 had a recurrence shortly after vaccination, and 5 remained NED (median follow-up of 26.4 months).
To determine the magnitude of T cell responses induced by the vaccine, post-vaccine PBMCs were stimulated with the peptide pools of full-length immunizing peptides (IMPs). Ex vivo immune responses were detected against these peptides in all 9 fully vaccinated patients. These responses were absent pre-vaccination and were sustained over time (up to 71 weeks after final vaccination). Ex vivo reactivity was detected against 66/105 (62.9%) IMPs, and no differences were detected between the two ipilimumab doses. By intracellular cytokine staining, both CD4+ and CD8+ T cell responses were observed, with CD8+ T cell responses observed in 66.7% of patients. IMP-specific T cells were polyfunctional.
The researchers assessed local dynamics at the vaccine and ipilimumab administration site by collecting fresh skin biopsies within 24 hours before and 48 hours after the third vaccination, and subjecting the biopsies to scRNAseq and scTCRseq. Clustering analysis revealed an increase in diverse myeloid and T/NK cell populations after treatment, with significant increases in various monocyte/macrophage populations, plasmacytoid dendritic cells (DCs), NK cells, and CD8 naive/central memory (Tcm) and effector memory (Tem) cells. Interestingly, both cDC1 and proliferating cDC2 were decreased, which could be due to dilution relative to the increase in other cells, or potentially suggesting trafficking to the lymph node.
Assessment of proinflammatory cytokine, chemokine, and effector function gene expression uncovered shifts of various cell types into more active phenotypes; genes related to cytokine and interleukin signaling, antigen processing and presentation, TLR stimulation, and IFN signaling were upregulated after vaccination.
Next, the T cell clonal dynamics were assessed by longitudinal assessment of the bulk circulating T cell TCRβ repertoire. T cells were collected before nivolumab treatment, after nivolumab and before vaccination, and after vaccination. In all patients, more TCRβ clonotypes significantly expanded after vaccination than after nivolumab alone. Among all clonotypes, besides clones present before treatment, hundreds of unique new clonotypes emerged both after nivolumab and after vaccination. The post-vaccine clonotypes reflected 7.6–25.9% of the unique clones in the circulating repertoire detected after vaccination, and 7.6–31.3% of these were significantly expanded after vaccination.
To determine if these newly emerged TCRβ clones were IMP-specific, expanding clones from IMP-stimulated PBMCs were subjected to TCRβ sequencing. The majority (median 92.9%) of these clones matched in sequence with the post-vaccine local clones. The IMP-expanded TCRβ clonotypes emerging after vaccination could be detected at later timepoints, representing 12.4-41.2% of post-vaccine clonotypes, while <0.3% of existing and post-nivolumab clones were IMP-expanded.
A post-treatment biopsy during the partial response phase from the patient who eventually achieved CR (P103), had an increase in the proportion of T cells and decrease in monocytes/macrophages, which was not observed in the 3 other patients with post-treatment biopsies. Using an algorithm (ProjecTILs) to phenotypically characterize the tumor-infiltrating lymphocytes, in the P103 sample, various CD8+ T cell phenotypes (Tem, precursor exhausted, and exhausted) expanded post-nivolumab and post-vaccination, while the other 3 patients had higher levels of CD4+ T cells.
In both P103 and another unresectable metastatic biopsy (P101, who was NED upon enrollment, but had a recurrence during vaccination and received palliative radiotherapy), new TCR clonotypes were detected after nivolumab, as well as a different set emerging after vaccination. However, in P101, the majority of the post-nivolumab clones were not detected after vaccination, while in P103 a large proportion persisted and/or expanded, with more novel clonotypes observed after vaccination. For P103, the overlap between intratumoral and significantly expanded peripheral TCRβ clonotypes was 44.6% post-nivolumab, while it was 9.2% pre-treatment. Compared to P101, the T cells from P103 after nivolumab or vaccination had higher levels of effector, inhibitory, and exhaustion marker expression.
The researchers then assessed whether these expanded clonotypes were vaccine neoantigen-specific by comparing the TCRβ sequences of the intratumoral T cells with those from the in vitro sequenced IMP-expanded clonotypes for each patient. This revealed an overlap of 7–29 clonotypes per patient, which represented 0.4–2.7% of the tumor TCRβ clonotypes. Comparing TCR sequences between tumoral and peripheral T cells after vaccination revealed 19–60 TCRαβ clonotypes that were shared in the 4 patients assessed. Of these, 96 TCRs were selected for antigen specificity testing. The TCRs were reconstructed in T cells from healthy donors, and their reactivity was tested against autologous B cell lines pulsed with immunizing neoepitopes. The specificity against the mutated vaccine neoepitope was confirmed in 35/96 (36%) of the TCRs. Almost all these TCRs were detected for the first time in the blood after vaccination.
The data in this study show that specific antitumor T cell responses can be triggered with the dual adjuvant-enhanced vaccine and ICB combination strategy. The responses and efficacy results in one patient are exciting, and larger trials will have to establish efficacy in a larger group of patients, as well as biomarkers for stratification of patients likely to respond.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first author Derin Keskin and lead author Patrick A. Ott answered our questions.

What was the most surprising finding of this study for you?
PAO: Conducting an investigator-initiated clinical trial in which a customized drug is designed and manufactured for each patient, and fresh tissue specimens are obtained from multiple organ sites is VERY labor-, cost-, and time-intensive. The academic setting provides a unique opportunity for such an endeavor, as each patient becomes “a labor of love”, given that the patient (who needed to go through multiple biopsies and leukaphereses procedures for our trial), the clinical investigators, and the laboratory scientists spent incredible amounts of time and dedication to enable the study treatments and the deep immune profiling performed on the study.
What is the outlook?
DK: There is no approved T cell-based vaccine formulation. The formula we used in this study was an improvement on our previous vaccine; however, T cell-based vaccines need a lot more work to reliably induce antigen-specific, high-avidity T cells with a long-term memory population.
PAO: We are quite encouraged by our observation that tweaks in the vaccine design can have substantial impact on their immunogenicity. More broadly, the results support the notion that technological advances in vaccine format and delivery can improve their ability to trigger anti-cancer immune responses. Next steps include further improvements in neoantigen target discovery and more effective combination with drugs addressing the suppressive tumor microenvironment. Hopefully at some point comparative studies will allow us “pick the winner” among different vaccine platforms. An ongoing phase 3 study is currently testing the personalized neoantigen vaccine.
Who or what has been a major source of inspiration or motivation for you throughout your career?
DK: Carl Sagan was the most significant influence in my early life to push me into science. His TV show and books were fascinating. I wonder how many people decided on a career in science due to Cosmos. I wanted to dedicate my PhD thesis to his memory and send a copy to his widow. I'm still sad that I had to graduate quickly and couldn't follow through with this plan. I was on a deadline and the thesis was not that great.
PAO: I did not appreciate early on in my career how important the role of a mentor is – particularly and unsurprisingly during the very early stages of a career. I have thought of (and sometimes reached out) to my PhD and post-doc mentors when I hit a major career milestone. Those “flashbacks” have come almost as a reflex – as if, in my mind, they are still overseeing my career.



