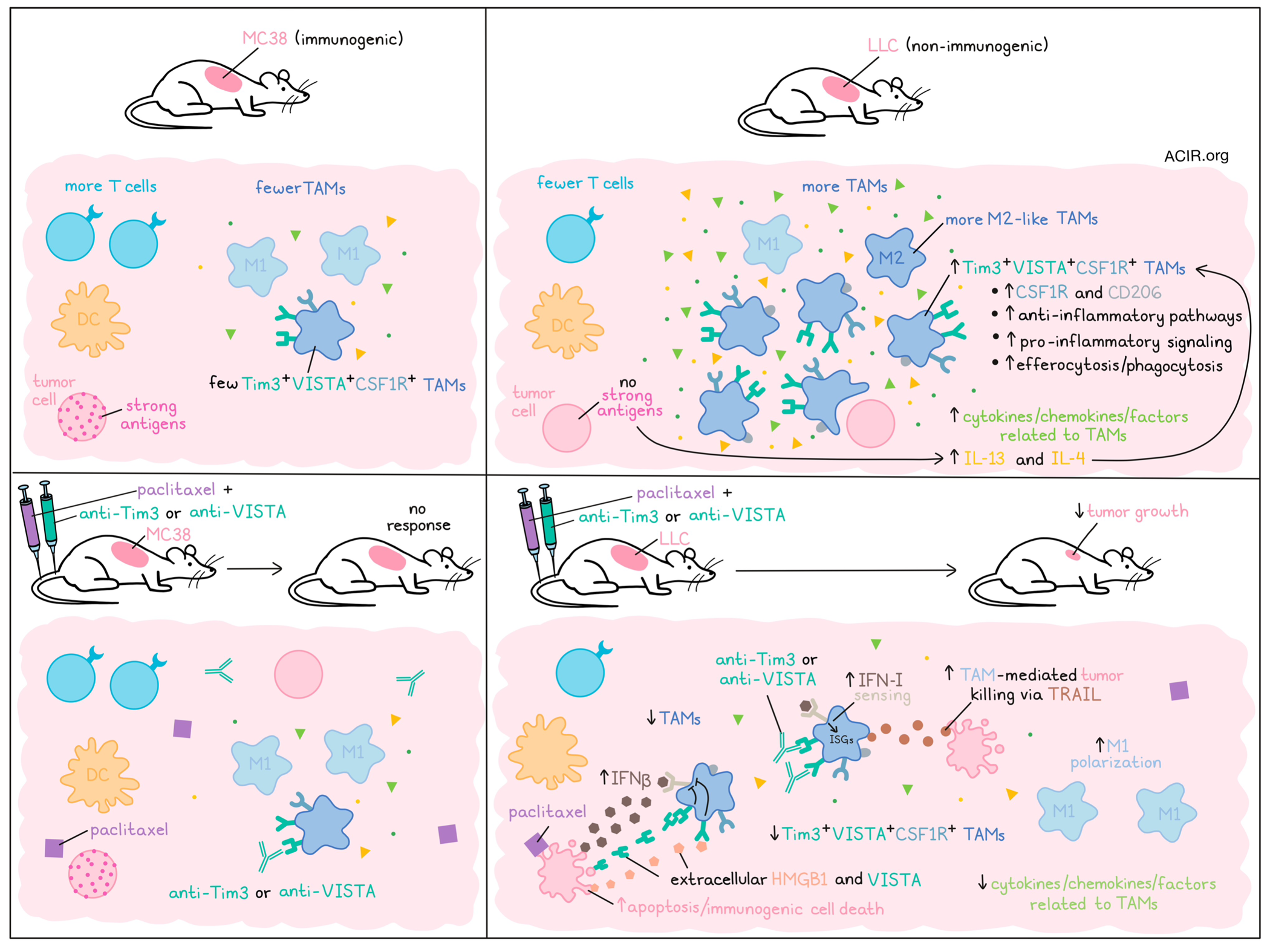
Immunosuppression by tumor-associated macrophages (TAMs) is thought to be one of the resistance mechanisms limiting the efficacy of immune checkpoint blockade (ICB) in cancer. In a recent publication in Science Advances, Vanmeerbeek et al. analyzed a newly described TAM subset present in unresponsive tumors, and assessed ways to target this population.
The researchers began by evaluating nine major inhibitory receptors on macrophages in human ICB-non-responsive tumors using single-cell RNAseq datasets. In melanoma, myeloid cells were enriched for the receptor genes HAVCR2 (encoding TIM3) and VSIR (encoding VISTA). In a multi-cancer dataset, this co-expression was also observed in TAMs, but not in monocytes, dendritic cells, T cells, B cells, or NK cells. The HAVCR2hiVISRhi phenotype was mostly associated with CSF1Rhi TAMs. A colorectal cancer dataset, including both immunogenic microsatellite-unstable and nonimmunogenic microsatellite-stable CRC, revealed that the HAVCR2hiVISRhi phenotype correlated with anti-inflammatory SPP1+ TAMs in non-immunogenic tumors, and was enriched in genes related to phagocytosis, pattern recognition receptor signaling, and anti-inflammatory signaling.
Further assessing this TAM phenotype, unbiased bioinformatics analysis was performed across macrophages obtained from various human scRNAseq studies, using 26 markers for different TAM subsets or polarization states. This revealed that HAVCR2hiVISRhi macrophages were a distinct subset, co-clustering with anti-inflammatory polarization markers such as CSF1R and CD163. This phenotype was present in tumors, but not healthy tissue, and was not observed in other disease states. TCGA data showed that the more immuno-resistant tumors correlated with the HAVCR2hiVISRhi TAM signature, which was associated with shorter overall and progression-free survival in various pan-cancer analyses.
The researchers moved to murine models to determine whether this subtype was also present in mouse tumors. Two models were used: the non-immunogenic Lewis lung carcinoma (LLC) model and the immunogenic MC38 model. Compared to the MC38 tumors, LLC tumors had high levels of TAMs and low levels of T cells in the tumor, with more anti-inflammatory M2-like TAMs. Tim3+VISTA+CSF1R+ TAMs were detected in both tumor types, but there were significantly more in LLC, and these TAMs had a higher expression of anti-inflammatory markers CSF1R and CD206.
scRNAseq data of LLC tumor-derived immune cells showed that the Havcr2+Vsir+ phenotype was only detected on TAMs, as in humans. A differential pathway enrichment analysis revealed that the Havcr2+Vsir+ TAMs were enriched for pathways related to anti-inflammatory activity, efferocytosis, and proinflammatory signaling. Furthermore, Tim3+VISTA+ TAMs were found to be hyperphagocytic in vitro.
To determine whether these TAMs could be specifically targeted to improve therapy outcomes, antibodies blocking Tim3 or VISTA were tested. Both LLC and MC38 tumors were resistant to this approach. Next, several TAM-relevant targets were targeted in LLC models, including SIRPα, CD40, CCL2, TGFβ, and IL-10, but again, tumors were resistant. Taking another approach, the researchers considered whether repolarization of these TAMs might be effective. Based on the enrichment of pathways related to apoptotic cell efferocytosis and pro-inflammatory signaling in Havcr2+Vsir+ TAMs, treatment with an immunogenic cell death (ICD)-inducing chemotherapy (paclitaxel) was assessed. While both tumor models were resistant to paclitaxel alone, the LLC tumors responded to the combination of paclitaxel and Tim3 or VISTA blockade. This synergistic approach reduced tumor growth, reduced Tim3+VISTA+CSF1R+ TAMs, and skewed TAMs to an M1-like phenotype.
In LLC tumors treated with the combination of paclitaxel and Tim3 or VISTA blockade, there was no increase in antitumor T cells or DC subsets. To confirm that modulation of TAMs was responsible for the efficacy, macrophages were depleted with clodronate liposomes. TAM depletion alone (no treatment) reduced tumor growth and made tumors susceptible to paclitaxel killing, while CD8+ T cell depletion had no impact. An enrichment of cytokines/chemokines/ factors related to TAMs was observed in untreated and paclitaxel-treated LLC tumors, which were depleted in tumors treated with the paclitaxel and Tim3/VISTA blockade combination.
LLC tumors treated with the combination therapy had increased apoptosis. To determine whether the TAMs were killing the tumor cells, TAMs were isolated from paclitaxel-treated LLC tumors and cultured with LLC cells. Blockade of various macrophage-associated toxicity pathways showed that TAMs caused tumor cell death, and this effect was disrupted when TNF-related apoptosis-inducing ligand (TRAIL) was blocked, implicating TAM-secreted TRAIL as the mechanism of cell killing. Further, in engineered mice with Havcr2-Visr- macrophages, LLC tumor growth slowed, and the addition of paclitaxel reduced tumor growth further.
In vitro experiments showed that a cytokine cocktail containing IL-4 was the most potent inducer of the Tim3+VISTA+ phenotype. In vivo, TAMs in LLC were more exposed to IL-4 and IL-13 than MC38 TAMs. This was related to a lack of strong antigens, as an OVA-LLC model had fewer Tim3+VISTA+ TAMs and a more immunogenic environment. Strikingly, the OVA-LLC model did not respond to the paclitaxel and Tim3/VISTA blockade combination.
The researchers then assessed the role of type I IFN sensing, since Ifnar1 was enriched in LCC, yet there was no enrichment of interferon-stimulated genes (ISGs) in LLC compared to MC38 tumors. LCC cells exposed to paclitaxel secreted IFNβ, a key mediator of immunogenic cell death-induced efficacy, but no clear ISG response was detected when the paclitaxel-treated LLCs were cocultured with macrophages. The ISG response recovered upon Tim3/VISTA blockade, suggesting the blockade reestablishes the IFNβ-induced ISGs in these TAMs. The researchers validated this using Ifnar-/- mice, in which the therapeutic effects of combination therapy were absent. Further, it was shown that extracellular HMGB1 and VISTA expressed by LLC cells treated with paclitaxel were the ligands engaged by Tim3 and VISTA, respectively (as VISTA exhibits homotypic interactions), to inhibit TAM response to type I IFNs.
Finally, the researchers validated these data in orthotopic-like tumor models, using the neoantigen-low YUMM1.7, OVA-expressing YUMM1.7, and neoantigen-high YUMMER1.7 melanoma models. YUMM1.7 had higher levels of TAMs and lower levels of T cells than the other two models. This model also had more Tim3+VISTA+CSF1R+ TAMs, more IL-4/IL-13 exposure to the TAMs, and was resistant to PD-1 blockade, while the other two models responded. All three models were resistant to paclitaxel monotherapy, but the combination of paclitaxel and Tim3/VISTA blockade synergized in the YUMM1.7 model only, with a decrease in Tim3+VISTA+ TAMs.
These data suggest that in immune-cold ICB-resistant tumors, Tim3+VISTA+ TAMs may play an important role in immunosuppression and therapy resistance. It will therefore be of interest to establish whether the combination of induction of immunogenic cell death and simultaneous VISTA or Tim3 blockade suggested here is effective in human tumors to repolarize the TAMs and sensitize tumors to therapy.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Isaure Vanmeerbeek and lead author Abhishek D. Garg answered our questions.

What was the most surprising finding of this study for you ?
This study was challenging because we were trying to study something exceptional i.e., two different immune- heckpoints (TIM3/VISTA) converging onto a similar immunosuppressive pathway in tumor-associated macrophages (TAMs), rather than exhausted T cells. Therapeutic targeting of these TAMs was a major obstacle, since it was not their depletion, but rather, their repolarization that was necessary to blunt tumor growth. During this research, we made at least three discoveries that really surprised us: (1) the specific association of TIM3+VISTA+ TAMs with neoantigen-low “T cell-cold” tumors in both humans and mice. This association was so strong that these TAMs disappeared if these tumors turned “T cell-hot”; (2) that these TAMs mediated direct anticancer cytotoxicity via TRAIL; and (3) that therapeutic activation of these TAMs could autonomously achieve tumor suppression without the need for anticancer T cells.
What is the outlook ?
Our results re-emphasize that the immuno-oncology field should rethink the depletion of TAMs as a therapeutic strategy. Our main conclusions join an emerging opinion that the, re-polarization of TAMs is more effective at overcoming immuno-resistant tumors. Herein, we come up with a novel strategy to achieve this, i.e., TIM3/VISTA blockade repositioned for exclusive treatment of TAM-enriching “T cell-cold” tumors in combination with immunogenic chemotherapy. Our results may explain why blockade of TIM3/VISTA has limited efficacy in some early clinical trials, especially in “T cell-hot” tumors (where they are expected to work due to their association with exhausted T cells). These observations may be important for the field, since overcoming “T cell-cold” tumors is the next big frontier for immuno-oncology.
What was the coolest thing you’ve learned (about) recently outside of work ?
ADG: I recently learned that paleontologists found a prehistoric site with fossils of animals that died within hours after a massive asteroid hit the earth and initiated the extinction of dinosaurs. They confirmed the presence of the asteroid impact’s debris in the gills of the fossilized fish. I thought this was really cool – a fossilized record of the aftermath of an asteroid hit!
IV: Already from a young age, I was always watching and helping my mom in the kitchen. Especially baking, kneading dough, or whisking batter were my favorite activities. Even now, I still have “baking Sunday” where I try out a new recipe. Recently, I learned via MasterChef that when baking blueberry pancakes, you should first pour the batter into the pan and only then add some blueberries and not mix them into the batter directly. So, this Sunday, I’ll be making my best blueberry pancakes!



