T cell function and infiltration are inhibited in the tumor microenvironment (TME), which can be challenging to overcome with therapeutic vaccination, and approaches to effective activation of T cells in situ are needed. Cytomegalovirus (CMV) causes predominantly asymptomatic infections and is highly prevalent in humans. T cell responses against CMV are long-lived, increase with age, and are functional in patients with cancer. As recently reported in PNAS, Çuburu et al. investigated intratumoral (i.t.) injection of CMV-derived peptide epitopes in various mouse models, aiming to therapeutically reactivate preexisting anti-CMV T cell responses and enhance antitumor responses.
Mouse CMV (MCMV) infection mimics human infection. To assess whether it also induces inflationary T cell responses (memory responses growing over time), the researchers evaluated CD8+ T cell responses against the inflationary IE3 and noninflationary m45 CMV epitopes. IE3-specific T cells increased between the first and fifth months after infection, while levels of m45-specific T cells were high and remained stable. The inflationary T cells had a terminally differentiated phenotype (CD127loCD62Llo), while the noninflationary T cells had a mixed effector memory/central memory phenotype (CD127hiCD62Llo/hi). Restimulation of blood with seven MHC-I CMV epitopes resulted in the production of IFNγ and limited TNFα, but no IL-2.
To assess the tissue distribution of inflationary and non-inflationary T cells in subcutaneous (s.c.) tumors, C57BL/6 mice were infected with MCMV and six months later inoculated s.c. with TC-1 tumor cells. By day 12, established tumors had high levels of both inflationary and noninflationary MCMV-specific CD8+ T cells, while there were no detectable levels of CMV DNA or RNA in tumors. Most T cells in the tumor expressed CD69 and did not express CD103, suggesting these T cells had differentiated into resident cells.
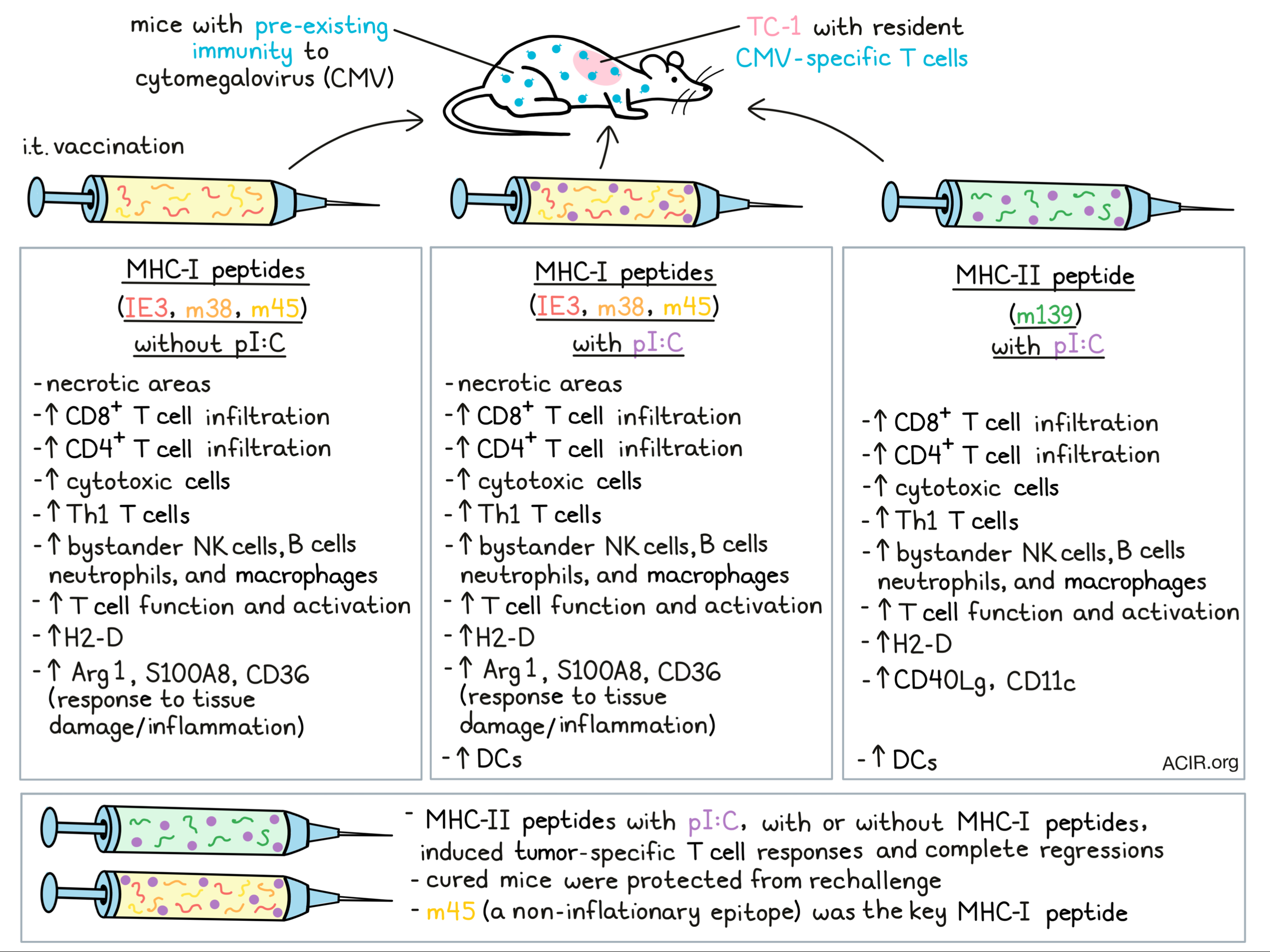
To assess whether these tumor-infiltrating MCMV-specific T cells could be activated, C57BL/6 mice were inoculated with TC-1 cells after MCMV infection, and established tumors were intratumorally (i.t.) injected with IE3, m38, and M45 MHC-I peptides, or m139 MHC-II peptide, with or without pI:C (a molecular adjuvant). Tumors were injected three times over a 6-day period, and two days after the last injection, tumors were harvested for analysis. Tumors that received MHC-I-restricted epitopes with or without pI:C had large necrotic areas and CD8+ and CD4+ T cell infiltration. Tumors treated with MHC-II peptides and pI:C also had CD4+ and CD8+ T cell infiltration, but lacked necrosis. Nanostring Immune Profiling of tumor tissue showed that injection of MHC-I peptides with or without pI:C and MHC-II peptides with pI:C resulted in increases in CD8+, cytotoxic, and Th1 cells, as well as bystander NK cells, B cells, neutrophils, and macrophages. In contrast, dendritic cells were only increased in groups treated with pI:C. Further assessment of the Nanostring panel for TME-associated genes revealed that treatment with MHC-I epitopes (with or without pI:C) or MHC-II epitopes (with pI:C) resulted in the upregulation of a high number of genes and limited downregulation of genes. Upregulation of genes related to T cell function and activation (T-bet, granzyme K, CXCR3, PD-1) was observed. In these tumors, there was also an increase in gene expression of H2-D, while CD40Lg and CD11c genes were increased in tumors treated with the MHC-II epitopes plus pI:C. MHC-I epitopes increased Arg1, S100A8, and CD36 genes, all involved in response to tissue damage or inflammation.
To assess whether these injections were therapeutic, TC-1 tumors (~100 mm3) in latently infected mice were injected six times over 2 weeks with IE3, m45, and m38 MHC-I epitopes with or without pI:C. These treatments resulted in delays in tumor growth and improved survival. Because the treatment resulted in acute, sometimes fatal toxicity in some mice, the researchers assessed lower doses (0.1 and 0.01 μg) and found that when combined with the adjuvant, toxicity did not appear, while survival was still improved.
To assess whether sequential injection of the MHC-I and MHC-II peptides could trigger immune activation and tumor killing, TC-1 tumors were injected six times with various combinations or in different orders of a mixture of MHC-I (IE3, m38, m45) or MHC-II (m139) peptides plus pI:C. All groups displayed tumor growth reduction, but complete regression was only observed in mice that received MHC-II peptides with or without MHC-I peptides. To determine whether treatment resulted in the induction of tumor antigen-specific T cells responses, CD8+ T cells specific for the TC-1 epitope HPV E7 were assessed in the blood of treated mice. All groups that had received MHC-II peptides had high levels of these tumor-specific T cells, at levels as high as 1-10% of the total CD8+ T cell population in mice treated with MHC-II peptides followed by MHC-I peptides. The levels of tumor-specific T cells in the blood correlated with complete tumor regression and long-term survival. When cured mice were rechallenged with tumor four months later, no palpable tumors developed, indicating induction of protective immunity.
To assess which epitopes contributed most to the tumor regression, TC-1 tumor-bearing mice were injected with one of the MHC-I epitopes with pI:C and the MHC-II peptide. When the noninflationary epitope m45 was injected, it resulted in similar results as the pool of peptides, and tumor control was better than in the other treatment groups.
Finally, the researchers assessed whether treatment with these CMV peptides also had therapeutic potential in the immunologically “cold” tumor, B16F10. Mice were treated with repeated i.t. injections of MHC-I- and MHC-II-restricted epitopes and pI:C. Treatment with m45 (MHC-I) together with m139 (MHC-II) peptides reduced tumor growth and prolonged survival. The treatment was associated with intratumoral infiltration of CD8+ T cells, m45-specific CD8+ T cells, and a limited increase in NK cells, while it decreased the levels of B cells, granulocytic cells, and CD4+ T cells. Treatment also increased the tumor concentration of IFNγ, CXCL9, CXCL10, CCL2, and CCL4, and increased blood cytokine and chemokine concentrations.
Overall, reactivating anti-CMV T cell responses in tumors was found to induce antitumor immune responses, even in immunologically cold tumor environments, resulting in tumor control in mouse models. This tumor antigen-agnostic approach, coupled with the widespread prior exposure to CMV in humans, suggests this could be a useful, off-the-shelf approach to immunotherapy using readily synthesizable epitope-length peptides in radiologically accessible tumors.
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, first author Nicolas Çuburu answered our questions.
What was the most surprising finding of this study for you?
We had three major surprises. First, repeated intratumoral injection of minimal CMV-derived peptide CD8+ T cell epitopes induced remarkable rapid and potent tumor regression. Second, a CMV peptide that was recognized by noninflationary CMV CD8+ T cells (that have more of a memory phenotype) was more effective than a peptide that was recognized by inflationary CMV CD8+ T cells (that have more of an effector phenotype). Third, a CD4-restricted CMV peptide, in the absence of CD8+ activating peptides, induced profound immune activation of the tumor microenvironment, leading to durable tumor regression in some treated animals, but only if co-injected with poly I:C.
What is the outlook?
As a new class of agent for in situ cancer therapies, cytomegalovirus-derived peptides will likely have to be evaluated in combination with established standard of care, such as immune checkpoint blockade, surgery, and/or chemotherapy. Choosing the right type of cancer and stage of progression for initial clinical evaluation will be key. While intratumoral drug delivery may represent a technical challenge for administration, this route of drug delivery has a potentially high therapeutic index and limited systemic toxicity. However, the peptides do not preferentially bind the tumor cells, and patients will vary in the size of their tumors and their preexisting CMV T cell immunity to the peptides. Therefore, peptide dosages may have to be individualized to limit adverse side effects. One possibility would be to do dose escalation in every patient until a dosage that induces a therapeutic response with minimal toxicity is reached. This approach should be possible because, unlike most biologics, the peptides should be too short to induce inactivating anti-drug antibodies.
What was the coolest thing you’ve learned (about) recently outside of work?
Recently I learned to sail Flying Scot, a 19-foot sailboat designed for lake sailing. It is very popular in the Chesapeake Bay, which is an hour away from where I live. It is a great hobby that I can do leisurely with my wife and 6-year-old daughter or competitively in regattas. I enjoy the technical, intuitive, and observational skills involved with sailing. For me being out on the water is perfect to unwind and recharge after a week in the lab.




