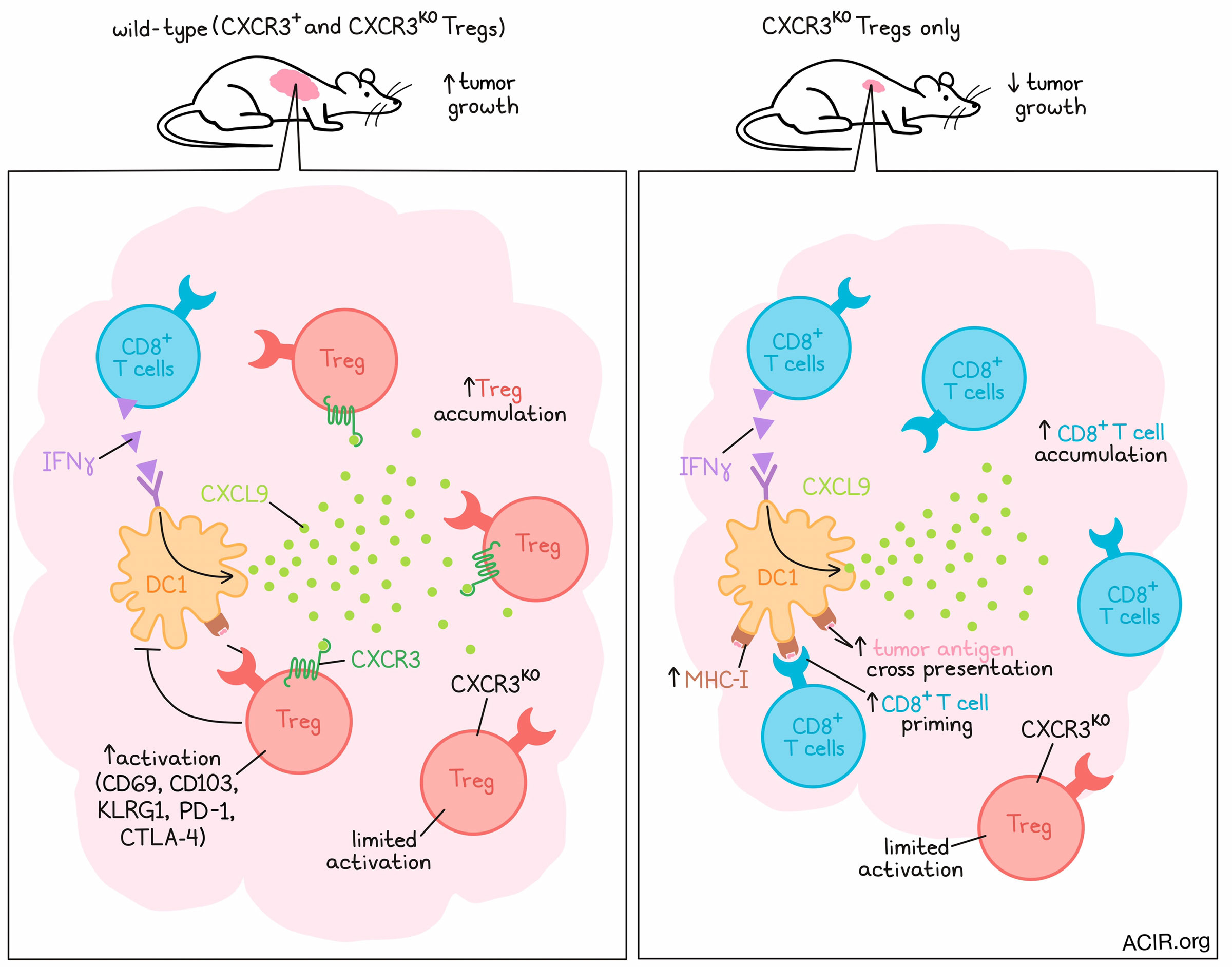
Regulatory T cells (Tregs) often present a challenge to antitumor immunity, but exactly how they suppress immune responses is not entirely understood. Investigating the accumulation and activation of Tregs in tumors, Moreno Ayala et al. found that CXCR3+ Tregs interacted preferentially with CXCL9+ DC1s in tumors, and that genetic knockout of CXCR3 specifically in Tregs enhanced DC1 cross-presentation of tumor antigens, CD8+ T cell priming, and antitumor immunity. Their results were recently published in Immunity.
To begin, Moreno Ayala et al. investigated T cell phenotypes in mice bearing MC38 colon carcinoma, EL4 thymic lymphoma, or 9464D neuroblastoma tumors. While the frequencies of Tregs and CD8+ T cells varied across tumors, there was consistently a positive correlation between the two populations. The researchers also noted that CXCR3 expression was increased on CD8+ T cells, effector CD4+ T cells, and Tregs in tumors and draining lymph nodes relative to healthy tissues and peripheral lymph nodes, respectively.
An evaluation of dynamics showed that peak CXCR3 expression on Tregs correlated with peak CXCR3+CD8+ T cell infiltration into tumors, at which point Tregs began accumulating in dLNs. At the peak of tumor infiltration, antigen-experienced (CD44+) CXCR3+ Tregs showed increased markers of activation and effector function (CD69, CD103, KLRG1, PD-1, and CTLA-4) compared to CD44+CXCR3- Tregs. Together these results suggest that CXCR3 expression supports enhanced accumulation and activation of Tregs in tumors.
To study the effects of CXCR3 on Tregs specifically, Moreno Ayala et al. generated genetic mouse models that take advantage of X chromosome inactivation in female mice. In these models, the mice contained heterogeneous populations of CXCR3+ and CXCR3KO Tregs, with the option to deplete one Treg population or another (depending on the model) with the administration of diphtheria toxin. In mice depleted of CXCR3+ Tregs (leaving only CXCR3KO Tregs), growth of MC38, EL4, and 9464D tumors was delayed compared to when CXCR3+ Tregs were left intact or CXCR3KO cells were depleted. This effect was not observed when a similar proportion of Tregs was depleted irrespective of CXCR3, suggesting that CXCR3+ Tregs specifically play a role in suppressing antitumor effects.
Investigating this role further, the researchers found that in the setting of CXCR3KO Tregs, CD8+ T cells, including a population of tumor antigen-specific CD8+ T cells, were increased in tumors across MC38, EL4, and 9464D models. CXCR3 deficiency in Tregs did not affect the activity of CD8+ T cells, as measured by IFNγ and TNFɑ production upon ex vivo restimulation, nor did it appear to affect PD-1 expression, suggesting that CXCR3+ Tregs limit CD8+ T cell accumulation, but not activation. Further, the addition of anti-PD-1 could be used to enhance antitumor efficacy. Looking at the accumulation of Tregs in tumors, the researchers noted a modest reduction in Treg frequency associated with CXCR3 deficiency. Similar effects were also observed in a genetically engineered mouse model of sarcomagenesis (non-transplantable), in which depletion of CXCR3+ T cells after the formation of sarcomas led to increased intratumoral CD8+ T cells and reduced Tregs.
Looking at the quality of CXCR3KO Tregs, the researchers noted reduced expression of PD-1 and CTLA-4 throughout MC38 tumor progression, indicative of a less activated phenotype. In a direct competitive setting in mice with both CXCR3+ and CXCR3KO Tregs intact, CXCR3KO Tregs made up a smaller proportion of Tregs in tumors and dLNs and expressed less PD-1 and CTLA-4 compared to CXCR3+ cells, suggesting again that CXCR3 contributes to preferential Treg accumulation and activation in tumor tissues. Going a step further, the researchers tested the accumulation of CXCR3+ Tregs directly by adoptively transferring ex vivo-activated and -differentiated Th1-like Tbet+ Tregs with or without CXCR3 expression. After transfer to MC38 tumor-bearing mice, CXCR3KO Tregs were enriched in dLNs, but reduced in tumors compared to WT Tregs 24 hours after transfer, suggesting that CXCR3 expression itself was critical for Treg accumulation in tumors.
Next, the researchers hypothesized that CXCR3 in Tregs may function by promoting Treg colocalization with and regulation of DCs, which are known to produce the CXCR3 ligands CXCL9 and CXCL10 in C57BL/6 mice. To study this, the researchers developed a fluorescently tagged mouse model that allowed them to visualize and quantify interactions between Tregs and CD11c+ DCs as well as between CD8+ T cells and DCs. This showed that CXCR3-deficient Tregs were less likely to interact with DCs than CXCR3-competent Tregs, while CD8+ T cells increased and were more likely to interact with DCs when Tregs were CXCR3-deficient. In a competitive setting with both CXCR3+ and CXCR3KO cells intact, these results for Treg–DC interactions were similar, but the effect on CD8–DC interactions was abrogated.
Looking at CXCL9 and CXCL10 production, the researchers found that CXCL9 was produced almost exclusively by DCs, and that the number of CXCL9+CD11c+ cells positively correlated with the number of wild-type Tregs. This positive correlation was weaker when CXCR3 was knocked out in Tregs, and was not observed with CXCL10, which was not exclusive to DCs. CXCL9 was reduced in the absence of CD8+ T cells or IFNγ, supporting a hypothesis that IFNγ production by CD8+ T cells may contribute to DC upregulation of CXCL9, which in turn recruits CXCR3+ Tregs. This hypothesis was further supported by adoptive transfer experiments, which showed that the enhanced accumulation of CXCR3+ Tregs in tumors was lost in DC1-deficient mice.
Finally, the researchers hypothesized that CXCR3+ Treg cells may regulate the capacity of DC1s to stimulate antitumor CD8+ T cell responses, and tested their hypothesis using MHC-1-deficient MC38 tumors expressing OVA, which allowed the researchers to measure CD8+ T cell responses to DC cross-presentation. This revealed a significant increase in OVA-specific CD8+ T cells within tumors and dLNs in the context of CXCR3KO Tregs, but not CXCR3+ Tregs. This effect was dependent on the presence of DC1s. Enhanced cross-presentation of tumor antigens by DC1s was further confirmed using antibodies that bind to SIINFEKL-loaded H-2Kb molecules.
Altogether, these results describe a mechanism by which IFNγ from CD8+ T cells enhances CXCL9 expression by DC1s, which recruits CXCR3+ Tregs. These CXCR3+ Tregs in turn increase CXCL9+ DC1s, but limit DC1 cross-presentation of tumor antigens, impeding priming of antitumor CD8+ T cells and limiting their control over cancer progression. CXCR3 knockout in Tregs enhanced DC1 cross-presentation, CD8+ T cells, and antitumor efficacy, suggesting that CXCR3 could serve as a potential target for cancer immunotherapy.
Write-up and image by Lauren Hitchings



