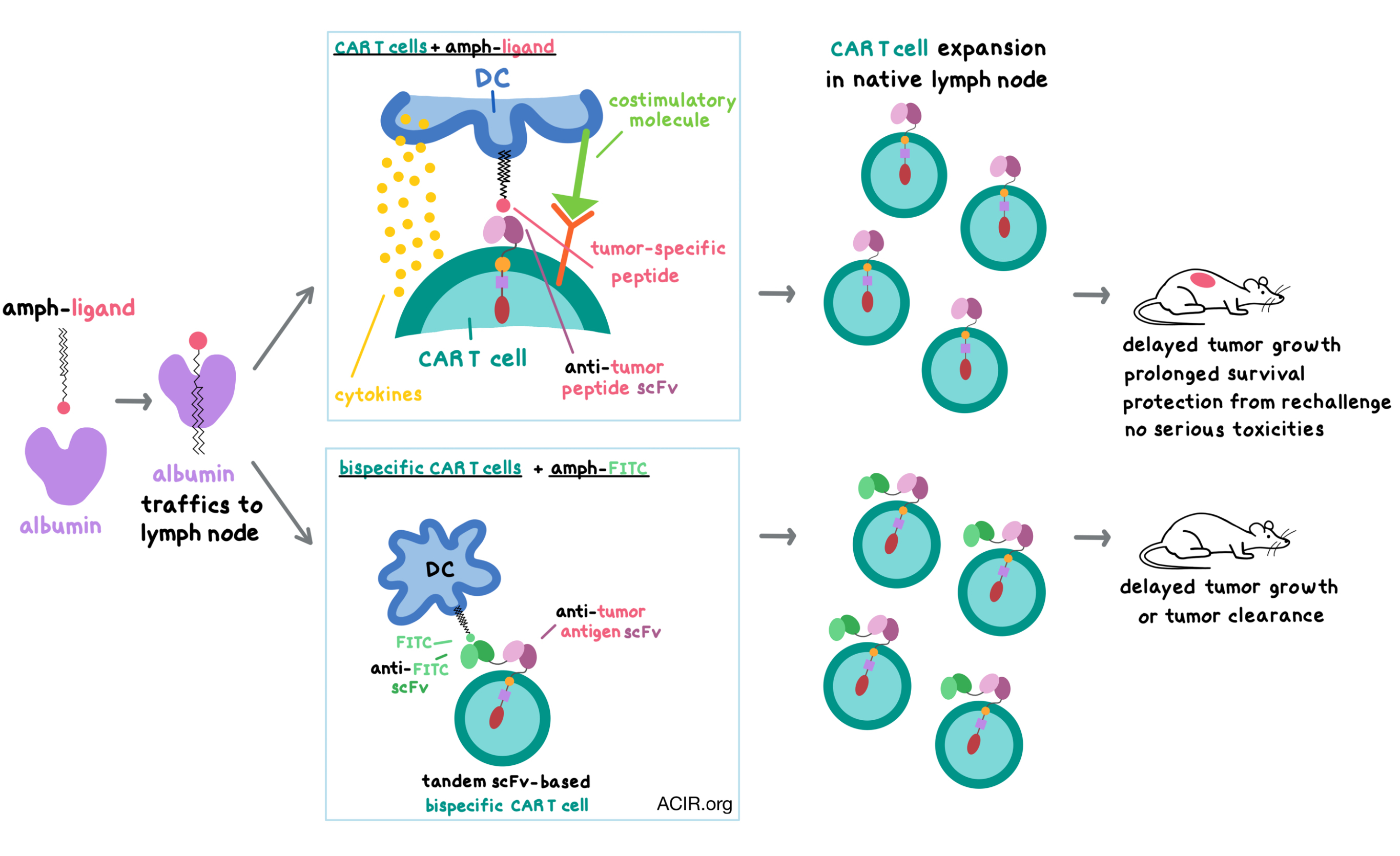
With the goal of improving the efficacy of CAR T cells in solid tumors and enhancing their functional persistence in all types of malignancies, Ma et al. developed a strategy to target CAR-detectable ligands to the lymph nodes in order to expand CAR T cells in the native lymph node environment. The results were recently published in Science.
To boost CAR T cells in vivo, Ma et al. created an amphiphilic CAR ligand (amph-ligand) by linking a small molecule or a peptide antigen to an albumin-binding phospholipid polymer; this design takes advantage of albumin’s natural propensity to traffic from blood to lymph (and therefore to lymph nodes), and of a lipid tail’s ability to insert into cell membranes, displaying the cargo. The authors hypothesized that the injected amph-ligands would bind to albumin, traffic to the lymph nodes, insert themselves into the membranes of antigen-presenting cells (APCs), and, together with the costimulatory molecules and cytokines produced by the APCs, prime the CAR T cells encountered in the lymph nodes.
Ma et al. first tested their hypothesis in vitro using amph-FITC. Upon incubation, amph-FITC was absorbed into the membrane of model APCs in a dose-dependent manner, and remained accessible on the surface despite continuous endocytosis, as indicated by anti-FITC staining. FITC-CAR T cells were stimulated by amph-FITC-coated target cells, resulting in target cell death.
Turning to in vivo testing, the team immunized C57BL/6 mice with amph-FITC and confirmed its localization to the draining lymph nodes. Within the lymph nodes, surface-accessible amph-FITC was preferentially found in CD11c+CD11b+ dendritic cells (DCs, which line the conduits carrying lymph to the lymph nodes) and macrophages. The duration of amph-FITC display on APC surface could be boosted by the STING agonist adjuvant cyclic-di-GMP; however, with or without the STING agonist, the amount of surface FITC decreased quickly and remained only on a small percentage of cells.
Having confirmed the ability of amph-FITC to traffic to the lymph nodes, the researchers examined its in vivo effects on CAR T cells. To do this, Ma et al. transferred CD45.1+ FITC-CAR T cells into lymphodepleted congenic CD45.2+ mice and then vaccinated twice either with amph-FITC alone or with amph-FITC plus adjuvant. Amph-FITC vaccination greatly expanded CAR T cells, and the expansion was further dramatically increased by the addition of the adjuvant. Phenotype analysis 3 weeks after vaccination revealed that the persisting CAR T cells were a mix of effector, effector memory, and central memory phenotypes. Similar results were observed in lympho-replete mice. A series of depletion experiments demonstrated that amph-FITC-mediated CAR T cell expansion was dependent on DCs and macrophages, but not Batf3+ DCs.
Next, the team evaluated the safety of amph-FITC immunization. Because only a small fraction of any cell type in the lymph nodes displayed detectable FITC ligand, the viability of lymph node cell populations was not affected. There were no changes in CAR T cell liver infiltration and serum cytokine levels, no liver toxicities were observed, and there was no antibody response against amph-FITC. The combination of CAR T cell transfer and amph-FITC vaccination did briefly reduce the ability to prime endogenous T cell responses, but it quickly recovered within 7 days of vaccination. Overall, the combination of CAR T cells and amph-FITC posed no serious safety concerns to mice.
To test the antitumor efficacy of this approach, the researchers created mouse CAR T cells expressing a CAR that recognizes an EGFRvIII-derived epitope, and synthesized an amph-pepvIII ligand utilizing the corresponding peptide with or without a FITC label. As expected, amph-pepvIII was inserted into cell membranes, stimulated the corresponding EGFRvIII-CAR T cells in vitro, and expanded CAR T cells in vivo. In lymphodepleted mice with EGFRvIII-expressing CT-2A glioma tumors, adoptive transfer of EGFRvIII-CAR T cells followed by amph-pepvIII immunization led to CAR T cell expansion in the periphery, increased proportion of effector CAR T cells, increased CAR T cell infiltration into the tumors, increased polyfunctionality, increased levels of granzyme B and Ki67, delayed tumor growth, and prolonged survival. Similar results were observed in non-lymphodepleted mice. Surviving vaccinated mice had persistent EGFRvIII-CAR T cells and were protected from rechallenge with both EGFRvIII-expressing CT-2A and parental CT-2A tumors, suggesting induction of an endogenous T cell response. This was confirmed by ELISPOT of splenocytes, which responded to parental CT-2A cells with strong IFNγ production when derived from mice that received CAR T cells plus amph-pepvIII, but not from mice that received CAR T cells only. In terms of safety, in both lymphodepleted and non-lymphodepleted mice, the combination of CAR T cells and amph-ligand boosting led to minimal changes in serum cytokine levels as well as small and transient changes in body weight.
Generalizing this amph-ligand boost strategy to any kind of CAR regardless of its cognate ligand, Ma et al. designed a tandem, scFv-based, bispecific CAR that bound both FITC and a tumor-associated antigen, thus enabling the CAR T cells to be activated in response to either molecule. To test this design, the researchers created a bispecific CAR recognizing FITC and melanoma-associated antigen TRP1. The FITC/TRP1-CAR T cells efficiently killed TRP1+ B16F10 target cells in vitro. In B16F10 tumor-bearing mice, amph-FITC immunization expanded CAR T cells in the periphery, increased their tumor infiltration, slowed tumor progression, and prolonged survival in a safe manner, whereas CAR T cells without amph-FITC boost had minimal effect on tumor growth. The efficacy of this system was similar between CD8+ CAR T cells and a mix of CD8+/CD4+ CAR T cells.
Next, the researchers tested the bispecific CAR/amph-ligand system in a metastatic tumor model. They created FITC/EGFRvIII-CAR T cells and transferred them into BALB/c mice bearing intravenously injected, EGFRvIII-expressing 4T1 tumors that had metastasized to the lung. CAR T cells alone did not greatly affect tumor progression, but the addition of an amph-FITC boost slowed tumor growth, prolonged survival, and even cleared tumors in 2 of 5 mice.
Aiming to confirm that the bispecific CAR/amph-ligand would be relevant in humans, the researchers generated FITC/CD19 human CAR T cells and demonstrated in vitro that both CD19+ Raji cells and amph-FITC-coated target cells stimulated the bispecific CAR T cells to secrete IFNγ.
Overall, Ma et al. designed and demonstrated a novel vaccine approach that enhances the number and functionality of CAR T cells in vivo and increases their efficacy against solid tumors in a safe manner. The combination of a bispecific CAR with amph-FITC immunization could provide a universal approach to enhancing CAR T cells that target any tumor antigen.
by Anna Scherer
Meet the researchers

This week, first author Leyuan Ma answered our questions.
What prompted you to tackle this research question?
CAR-T therapy demonstrates great clinical efficacy in treating leukemia. However, the time-consuming and costly manufacturing, as well as its lack of efficacy in solid tumors, restrains its broad implementation. Cancer vaccine, on the other hand, has huge potential in priming and amplifying highly active tumor-targeting T cells in the short term at very low cost, but it is restricted by the pairing of vaccine molecules and certain HLA molecules, which vary among people. It would be extremely helpful to cancer patients if we can devise such a vaccine that bypasses this HLA-restriction for CAR-T cells. We employed a lymph-node targeting “amphiphile vaccine” technology to precisely deliver the epitope recognized by the CAR into lymph nodes. On arrival, the epitope can be displayed on the surface of natural antigen presenting cells and presented to CAR-T cells through the direct interaction with the chimeric antigen receptor. This CAR-T-specific vaccination strategy has all the beneficial effects of conventional vaccine and is not restricted by the HLA molecules, making it available to every patient who is to receive CAR-T therapy.
What was the most surprising finding of this study for you?
The most surprising finding for me, and a qualitative distinction between “amphiphile vaccine” boosted CAR-T and conventional CAR-T cells, is that the “amphiphile vaccine” boosted CAR-T cells trigger pronounced endogenous immune response (T cells response in particular) in the host. It establishes long-lasting antitumor immunity in the host and prevents tumor escape caused by the loss of primary tumor antigen during CAR-T therapy. This can be a major reason why the "amphiphile vaccine” boosted CAR-T cells are highly effective against solid tumors.
What was the coolest thing you’ve learned (about) recently outside of the lab?
The coolest thing I have learned recently is scuba diving, to a depth of 30 meters in the ocean of the Philippines. It is the most amazing experience ever swimming inside a huge swarm of Jack fish.



