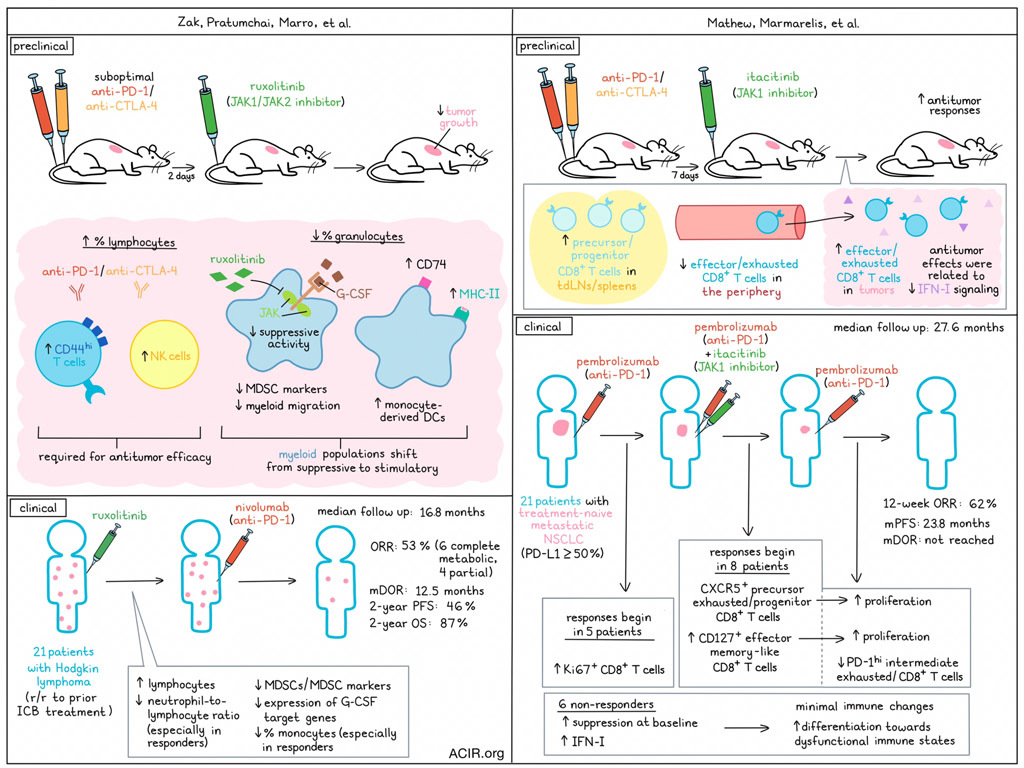
One of the main outstanding issues for cancer immunotherapy is how to improve the efficacy of immune checkpoint blockade (ICB) in subsets of patients who currently do not benefit. Two recent back-to-back publications in Science tackled this issue by combining ICB with JAK inhibition. Zak, Pratumchai, Marro, et al. performed preclinical and clinical studies in solid tumor models and Hodgkin lymphoma, while Mathew, Marmarelis, et al. assessed this strategy in preclinical models and patients with non-small cell lung cancer (NSCLC). Both studies assessed the immune changes induced by the combination treatment to assess its mechanisms of action.
Zak, Pratumchai, Marro, et al. focused on the JAK1/JAK2 inhibitor ruxolitinib after identifying JAK inhibitors as active in a screen of small molecules impacting T cell exhaustion in the chronic LCMV model. The murine MC38 model was treated with suboptimal doses of anti-PD-1 and anti-CTLA-4, as well as ruxolitinib starting 2 days after the first ICB dose. While ruxolitinib therapy or ICB therapy alone had limited impact on tumor growth, the combination significantly reduced tumor growth. Similar results were found in a B cell lymphoma and lung cancer model. Assessing the tumor immune environment in the treated MC38 tumors revealed that the combination therapy increased the percentage of lymphocytes and lowered that of granulocytes, compared to ICB alone. This resulted in higher numbers of CD44hi T and NK cells, and depletion experiments revealed that both CD8+ T cells and NK cells were required for the efficacy of the combination treatment.
Tumor-infiltrating granulocytes in the combination treatment group had reduced expression of myeloid-derived suppressor cell (MDSC) markers and higher expression of MHC-II molecules and Cd74. To further assess the contribution of these granulocytes, Ly6G+ cells were depleted during ICB treatment, which resulted in similar effects as ruxolitinib + ICB treatment, while Ly6G depletion did not further improve ruxolitinib + ICB treatment.
The researchers then assessed how ruxolitinib impacted tumor-infiltrating myeloid cells in three different murine tumor types. Cite-Seq of TILs and UMAP clustering revealed a lower percentage of myeloid cells resembling ARG1+ macrophages or monocytes (corresponding to TREM2+ macrophages) than in the ICB alone group. Additionally there was a lower percentage of a cluster with granulocytes in the ruxolitinib + ICB group, as compared to ICB alone. These clusters had the highest expression of MDSC markers. Two clusters with monocyte-derived DCs increased in frequency, suggesting a shift from suppressive to immune-stimulating myeloid populations.
Since suppressive activity of myeloid cells can be induced by excessive stimulation of G-CSF and GM-CSF, which signal through JAK kinases, the researchers assessed whether ruxolitinib treatment inhibited this suppressive programming of myeloid cells. An increase in the proportion of differentiated neutrophils was observed in the bone marrow of ruxolitinib and combination therapy-treated mice. Further, MDSC markers were downregulated in neutrophils in both the bone marrow and blood, and the expression of myeloid migration genes decreased in neutrophils in mice treated with ruxolitinib. The downregulated genes after treatment were induced by G-CSF, and the combination of ICB and anti-G-CSF limited tumor growth, suggesting G-CSF has immune-suppressive effects in this model.
Mathew, Marmarelis, et al. also performed preclinical research in which they investigated the JAK1 selective inhibitor itacitinib in combination with ICB. In the Res 499 model, a known ICB-resistant model derived from B16F10, treatment with itacitinib 7 days after ICB initiation resulted in improved antitumor responses. Comparing the effects of itacitinib with anti-IFNAR1 treatment showed similar effects, suggesting that the effects of itacitinib are related to reduced IFN-I signaling.
An assessment of changes in non-naive Ki67+CD8+ T cell responses after treatment showed a population of memory precursors and/or progenitor-like CD8+ cells (TPRE/PROG-like ) that increased in the draining lymph node and spleen after treatment. Additionally, an intermediate or circulatory subset of exhausted cells with effector-like features: CD8+PD-1hiTEX-INT-like cells, were found to decrease in the periphery and increase in the tumor after the combination therapy.
Moving to clinical research, Zak, Pratumchai, Marro et al. performed a Phase 1 clinical trial in patients with Hodgkin lymphoma in whom ICB treatment had previously failed. Patients were treated with ruxolitinib followed by nivolumab. Twenty-one patients were enrolled and 19 were evaluable for response. No dose-limiting toxicities were observed. With a median follow-up time of 16.8 months, the best overall response rate (ORR) was 53% (6 complete metabolic responses and 4 partial remissions). The median duration of response (DOR) was 12.5 months, and the 2-year progression-free survival (PFS) and overall survival (OS) were 46% and 87%, respectively.
Assessing PBMCs obtained at various time points during treatment revealed that the number of lymphocytes increased after ruxolitinib treatment. The neutrophil-to-lymphocyte ratio (NLR), which has previously been correlated to MDSC frequency and poor prognosis in cancer, was significantly lower after ruxolitinib treatment. Further, MDSC markers decreased after ruxolitinib treatment, as well as the percentage of polymorphonuclear MDSCs in the blood and the expression of G-CSF target genes. Comparing responders and nonresponders showed that complete responders had a greater reduction in the NLR and proportion of monocytes. Further, responders had more downregulation of a monocyte-enriched cluster than other groups. Therefore, treatment with ruxolitinib reduced the percentage of monocytes in the blood and the expression of suppressive genes in the monocytes.
Mathew, Marmarelis, et al. conducted a Phase 2 trial of pembrolizumab, followed by combination treatment with itacitinib, followed by pembrolizumab monotherapy in 21 patients with treatment-naïve metastatic NSCLC (tumor PD-L1 ≥50%). The 12-week ORR was 62%, and at a median follow-up time of 27.6 months, the median PFS was 23.8 months, and median DOR was not reached. Five patients had an early response to pembrolizumab before itacitinib was added (aPD1.R), while eight patients did not respond to pembrolizumab, but responded when itacitinib was added (JAKi.R), and six patients were nonresponders at week 12 (NR).
The researchers assessed CD8+ T cell features that changed after the initial pembrolizumab treatment, during combination treatment, and after return to pembrolizumab monotherapy. In the aPD1.R group, initial pembrolizumab treatment resulted in an increase in Ki67+CD8+ T cells. In the JAKi.R group, this early effect was not seen, but this group had an increase in CXCR5+ and CD127+ CD8+ T cells during combination treatment, followed by an increase in Ki67+CD8+ T cells after ending itacitinib treatment and continuing on pembrolizumab.
Looking further into the changes in the Ki67+CD8+ T cell population, in the JAKi.R group, the proportion of proliferating CD127+ TEFF-MEM-like and PD-1+CXCR5+TPRE/PROG-like cells increased, whereas that of PD-1hiTEX-INT-like cells decreased during itacitinib treatment. Assessing clonotype expansion and TCR sharing between these populations revealed that the JAKi.R group had an increase in the TPRE/PROG-like clonotypes and clonality that was not observed in the other groups after JAKi. These CD8+ T cells had a high degree of developmental relatedness with multiple different subtypes, suggesting fate-flexible properties.
The NR group had minimal changes in the CD8+ T cell compartment. This group had higher plasma levels of suppressive cytokines and pathways at baseline. PBMCs had high expression of signaling pathways related to IFN-I and suppressive cytokine pathways. Correlation analysis revealed a positive correlation between IFN-I signaling and progressive differentiation of CD8+ T cells toward terminal dysfunctional states, which may have negatively impacted response to therapy.
In summary, the two research studies presented here show the unexpected benefit of adding JAK inhibitors to ICB therapy to improve efficacy in various cancer types. Therapy affected myeloid cells and T cells in groups of patients, resulting in a modulation of the tumor immune microenvironment. These responses resulted in more patients benefiting from ICB. Further assessments will have to establish how to select patients who are most likely to benefit from this combination.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first author on “JAK inhibition enhances checkpoint blockade immunotherapy in patients with Hodgkin lymphoma” Jerry Zak answered our questions.

What was the most surprising finding of this study for you?
The most surprising finding is that an immune-suppressive drug used to treat autoimmune disorders is immune-enhancing when combined with checkpoint inhibitors. This counterintuitive observation suggests that even though the JAK–STAT pathway controls immune function across many tissues and cell types, the impact of blocking it varies depending on context, which can be therapeutically exploited to help patients with cancer and viral disease. For example, in both cancer and viral disease, we want to limit inflammation without preventing T cell responses to cancer and virus-associated antigens. We learned that in both experimental models and human patients, JAK inhibitors achieve this, although dosing, timing, and drug combinations are important.
What is the outlook?
We hope this paper will encourage more research on small molecules in immunotherapy and on myeloid-targeted immunotherapies. JAK inhibitors are not exclusively targeting myeloid cells, but myeloid modulation is crucial for their efficacy in combination with checkpoint inhibitors. This could be an example of a more general approach of therapeutically shaping myeloid cells to maximize the immune response to checkpoint inhibitors (or other T cell-targeted immunotherapies).
As next steps, patients are being enrolled for a phase II trial of ruxolitinib with nivolumab in anti-PD-1-relapsed or -refractory Hodgkin lymphoma. Hopefully, positive results in the phase II trial will help add this combination therapy to treatment guidelines for Hodgkin lymphoma. We are also actively developing clinical trials in solid tumors based on the positive results in lung cancer and the mechanistic studies pointing to a more general mechanism of action.
What was the coolest thing you’ve learned (about) recently outside of work?
I was in awe about the story of Nims Purja, a Nepalese climber who conquered all 14 eight-thousanders (mountain peaks above 8,000 meters above sea level) within 92 days. Moreover, he was part of the first successful team to summit K2 during winter. He is not exactly humble about it, but watching him go through this expedition in the Netflix documentary 14 Peaks: Nothing is Impossible with all the incredible mountain views was amazing to me.



