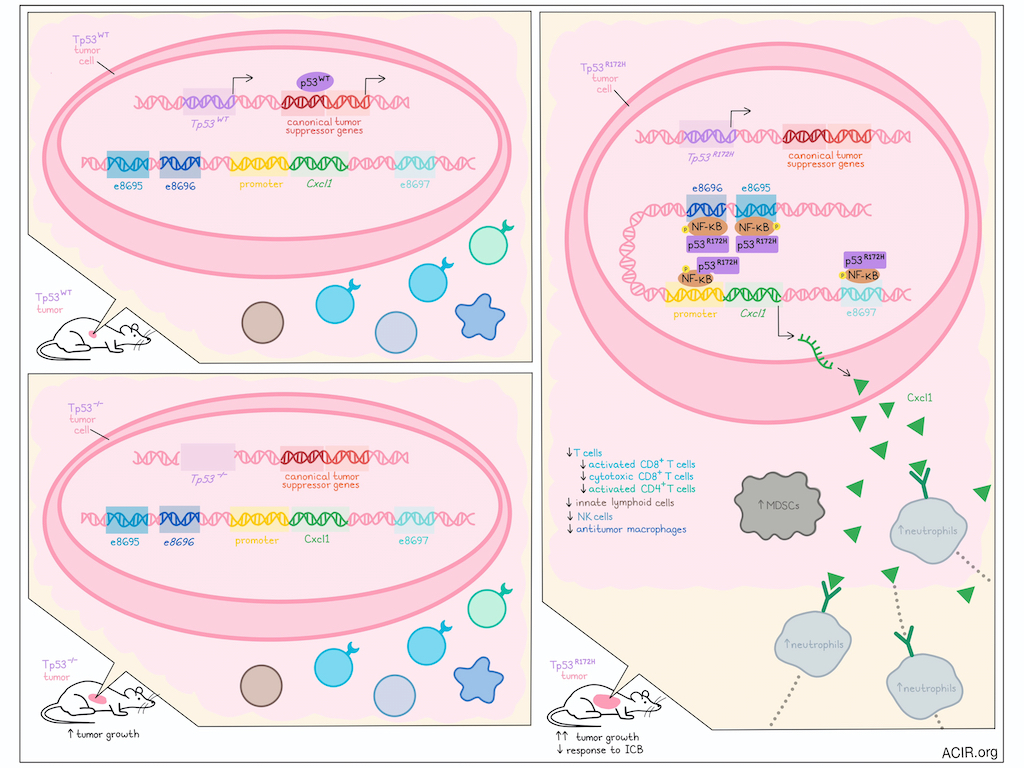
Mutations in the p53 gene are well known drivers of cancer due to the loss of tumor suppressor functions that are maintained by wild-type p53. However, in recent investigations, Mahat et al. found that a common mutant p53, p53R175H in humans and p53R172H in mice, also gained functions that supported the development of immunosuppressive tumor microenvironments, like those observed in pancreatic ductal adenocarcinoma (PDAC). Their work, recently published in Immunity, showed that mutant p53 maintained its transactivating activity and bound to enhancers of Cxcl1, driving Cxcl1 secretion and subsequently, recruitment of immunosuppressive neutrophils and MDSCs that supported tumor progression.
To study potential gains of function of mutant p53, Mahat et al. utilized KPC cells derived from a genetically engineered mouse model of PDAC, engineered with mutant Kras (present in the majority of PDACs) and mutant Trp53R172H (the murine equivalent of the common human genetic variant, Trp53R175H). In the presence of the mutant p53 allele, these cells lost expression of wild-type p53 (Trp53R172H/-), as observed previously, allowing for clear comparisons between Trp53R172H/- and isogenic Trp53−/− cells. Using RNAseq, the researchers identified several hundred genes under the control of p53R172H, with upregulated genes enriched for targets of NF-κB, and for various chemokines, including Cxcl1, Cxcl5, and Ccl2. The increased secretion of chemokines was confirmed in chemokine arrays, and ectopic expression of Trp53R172H cDNA in Trp53−/− cells partially restored chemokine expression.
Next, the researchers evaluated the tumor growth capacities of Trp53R172H/- and Trp53−/− cells by orthotopically implanting them into wild-type mice. After 21 days of growth, tumors formed by the Trp53R172H/− cells were significantly larger, less infiltrated with T cells (including activated and cytotoxic CD8+ T cells and activated conventional CD4+ T cells) and more infiltrated with neutrophils and MDSCs compared to tumors derived from Trp53−/− cells. Trp53R172H/− tumors were also less sensitive to immune checkpoint blockade (ICB; anti-CTLA-4 + anti-PD-1), suggesting that p53R172H likely supports PDAC growth by contributing to an immunosuppressive TME.
To establish whether p53R172H-dependent chemokines contributed to the immunosuppressive TMEs, the researchers established isogenic Trp53R172H/- cells lacking expression of Cxcl1, Cxcl2, or Cxcl5. Upon orthotopic implantation into mice, only tumor cells lacking Cxcl1 showed reduced tumor growth, with mice exhibiting better survival rates, which were further enhanced upon treatment with ICB. The TMEs in Cxcl1-deficient tumors were enriched in T cells, activated CD8+ T cells, cytotoxic CD8+ T cells, and activated conventional CD4+ T cells, recapitulating the TMEs in Trp53−/− tumors. Trp53R172H/- tumors lacking Cxcl1 also showed reduced neutrophils (which express Cxcr2 – the primary receptor for Cxcl1) and MDSCs (which can differentiate from neutrophils in the TME), and increased innate lymphoid cells, NK cells, and antitumor macrophages. Together, these results suggested that p53R172H-driven Cxcl1 expression supported the recruitment of suppressive immune cells and the development of an immune-cold TME.
Considering that mutant p53R172H loses its canonical DNA binding capacity, but maintains its transactivating domain, Mahat et al. investigated whether p53R172H might bind to new genomic sites. Using CUT&RUN, the researchers observed significant p53R172H occupancy at the Cxcl1 promoter, as well as at regions upstream up the Cxcl1 gene that were identified as probable enhancers of Cxcl1. Three enhancers (e8695, e8696, and e8697) were identified within 15 kb of the Cxcl1 gene. Deletion or ectopic expression of p53R172H respectively reduced or restored enhancer RNA transcription activity, suggesting that p53R172H binding at these enhancers likely influenced the transcription output of target genes. The researchers also confirmed the specificity of p53R172H occupancy and demonstrated that it was not the result of differential chromatin modeling. Further, using public datasets, the researchers found that p53WT occupied canonical targets, but showed no evidence of binding at the Cxcl1 promoter or enhancers, while p53R172H showed the opposite binding pattern, representing a unique gain of function.
To test which Cxcl1 enhancers supported Cxcl1 expression and to what extent, the researchers deleted e8695, e8696, and 8697 from Trp53R172H/- cells individually and in pairs. Deletion of e8695 or e8696 (but not e8697) significantly reduced Cxcl1 expression, with dual deletion modestly reducing Cxcl1 expression further. Tumors lacking e8695 and/or e8696 resembled those lacking Cxcl1, with TMEs showing increased immune infiltration and lower proportions of neutrophils and MDSCs, tumors showing slower growth, and mice surviving longer and responding better to ICB.
Given that the R172H mutation alters p53’s conformation and abolishes its canonical DNA-binding activity, the researchers hypothesized that p53R172H may be recruited to the Cxcl1 enhancers through interactions with other transcription factors. Analysis of the DNA sequences under p53R172H CUT&RUN peaks revealed no enrichment of the canonical WT p53 DNA-binding motifs or novel motifs, but aligned strongly with the NF-κB motif. Additionally, NF-κB showed increased phosphorylation and occupation of the Cxcl1 promoter and enhancers in Trp53R172H/− versus Trp53-/- cells, showing strong overlap with p53R172H occupancy. When NF-κB was inhibited using TPCA-1, binding of both NF-κB and p53R172H to Cxcl1 enhancers was reduced, as was Cxcl1 expression, suggesting that p53R172H binding was dependent on the NF-κB pathway.
To evaluate the relevance of their findings in human PDAC, Mahat et al. analyzed a pancreatic adenocarcinoma dataset from TCGA, and found that patients with high expression of the murine p53R172H-upregulated gene signature had significantly worse survival than those with low expression. Further, patients with DNA-binding-domain-mutant P53 had elevated levels of activated NF-κB and worse outcomes than patients with missense mutations predicted to undergo nonsense-mediated decay (P53-null tumors). Finally, the researchers identified human homologs of the mouse enhancers e8695, e8696, and e8697, and found that they were located within 15kb upstream of the CXCL2 gene (mirroring the proximity of e8695 and e8696 to murine Cxcl1). A strong NF-κB binding motif was identified in the human region homologous to e8695. Treatment with TNFα (to induce NF-κB translocation) supported NF-κB and p53R273H (but not p53WT) binding to the human enhancers, evidencing their functionality.
Together these findings suggest that mutant p53R172H not only loses the functionality of p53WT, but gains functionality by binding to enhancers of Cxcl1, facilitated by NF-κB, increasing Cxcl1 secretion and driving recruitment of immunosuppressive neutrophils and MDSCs that support an immunosuppressive TME. This data both suggests a novel mechanism by which p53 mutations drive tumor growth, but also provides insights into the potential for blocking Cxcl1 in PDAC to limit immunosuppression and support the efficacy of ICB.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Dig Bijay Mahat answered our questions.

What was the most surprising finding of this study for you?
The most surprising finding of this study was that mutant p53 reprograms the tumor microenvironment through enhancer-mediated gene regulation. Traditionally, mutant p53’s oncogenic gain-of-functions were thought to act mainly within tumor cells – driving proliferation, invasion, and survival. However, this study revealed an unexpected, cell-extrinsic role: by using distal enhancers, mutant p53 upregulates immunosuppressive chemokines, which recruit neutrophils and exclude T cells, thereby weakening responses to immune checkpoint therapy. This previously unrecognized enhancer-driven mechanism of immune evasion highlights an overlooked aspect of mutant p53 biology. It challenges the notion that p53 mutations are cell-autonomous, and suggests new therapeutic opportunities to target enhancers or chemokine pathways to enhance immunotherapy response.
What is the outlook?
This study redefines mutant p53 as both a tumor-intrinsic driver and a key modulator of the immune microenvironment, opening an exciting new frontier by linking mutant p53’s gain-of-function to immune evasion. Still, targeting enhancers in cancer cells –especially in dense solid tumors like pancreatic cancer – remains difficult. However, if enhancer-mediated regulation of chemokines, cytokines, or checkpoint inhibitors can be identified in immune cells, these enhancers might offer more accessible and effective therapeutic targets. Evidence suggests that targeting enhancers may be a safer approach than gene-targeting therapies. Future research should focus on mapping enhancer networks and transcription factors in immune cells, as well as assessing their effects on immunomodulation and immunotherapy outcomes in vivo.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
This collaboration, spanning three institutes and four labs, began with a simple hallway chat over coffee. If I could advise my early-career self, I’d say: never underestimate the power of relationships and resilience. Science is as much about navigating people and setbacks with patience and clarity as it is about ideas. We discarded over 90% of our initial data on gene expression comparisons between syngeneic p53-null cells after realizing they were confounded by divergent genomic aberrations arising from independently evolved genome instability in the absence of wild-type p53. Resisting the sunk cost fallacy, we went back to the drawing board, built isogenic models, recollected data with proper controls, and uncovered insights into this deeply studied, yet still mysterious biology.



