4-1BB plays an important role in the costimulation of T cells and is an attractive target for immunotherapy, however, clinical targeting of 4-1BB using agonistic antibodies has been plagued by either low potency or off-target liver toxicity due to Fcγ receptor (FcγR)-mediated cross-linking. To overcome these challenges and induce tumor-localized T cell costimulation, Claus, Ferrara, and Xu et al. designed a novel fusion protein that mediates 4-1BB agonism and tumor antigen targeting, without relying on Fc-mediated hyperclustering to induce 4-1BB activation. Their results were recently published in Science Translational Medicine.
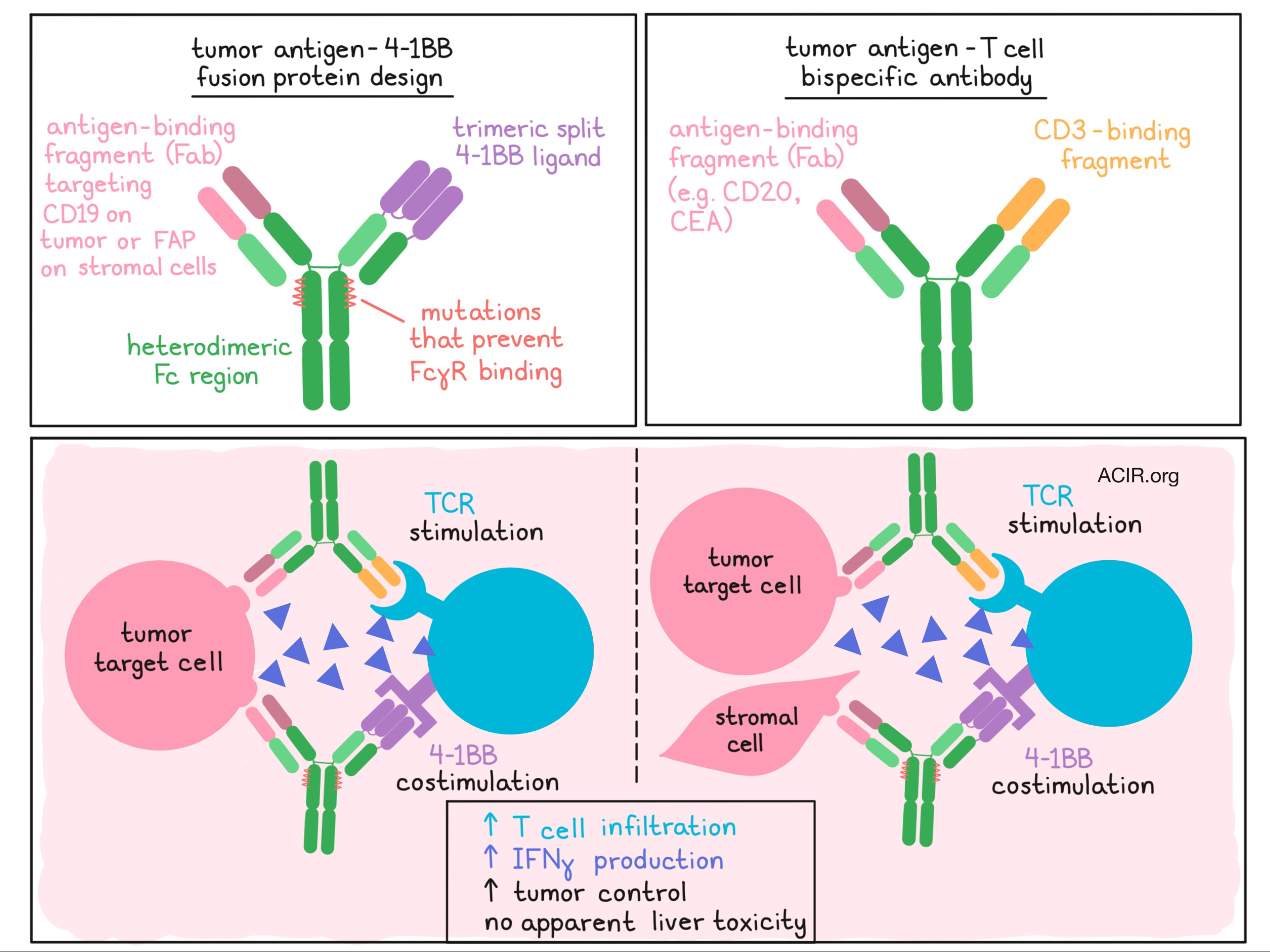
Claus, Ferrara, and Xu et al. designed a fusion protein consisting of:
- a trimeric split 4-1BB ligand (4-1BBL) to target 4-1BB
- a monovalent antigen-binding fragment (Fab) to target a tumor-associated antigen (either FAP or CD19)
- a heterodimeric Fc region containing mutations that inhibit FcγR binding but retain FcRn binding.
The design of the Fc region of this fusion protein allowed for IgG-like pharmacokinetics to be maintained (preventing rapid clearance of the construct from the tumor), while preventing the Fc-mediated cross-linking that has been associated with hepatic toxicity. Validating their design, the researchers showed in vitro that their fusion protein could simultaneously bind human 4-1BB and target antigen.
To test the efficacy of their fusion proteins, Claus, Ferrara, and Xu et al. began by testing the functionality of FAP-4-1BBL in vitro. In a coculture of 4-1BB+ human reporter cells and FAP-expressing cells, FAP-4-1BBL increased NF-kB activation. In cocultures of FAP-expressing cells and human PBMCs treated with agonistic anti-CD3 to stimulate TCR signaling, FAP-4-1BBL induced T cell activation, proliferation, and expression of costimulatory molecules without the addition of FcγR-expressing cells. Further, FAP-4-1BBL increased the number of central and effector memory T cells, increased the proportion of effector memory T cells, and increased effector memory CD8+ T cell production of IFNγ.
To determine whether FAP-4-1BB would induce similar effects on T cells in a tumor setting, the researchers used a patient-derived tumor digest of FAP-expressing epithelial ovarian cancer (EOC) plus CD3 stimulation. Here, FAP-4-1BBL did not increase proliferation or expression of infiltrating T cells, but did significantly increase IFNγ production by CD8+ T cells. In various patient-derived melanoma digests stimulated with CD3, FAP-4-1BBL increased inflammatory cytokines, but no clear patterns of cytokine modulation were identified between samples.
To test whether natural FAP expression in tumors was sufficient to provide functional cross-linking, the researchers combined FAP-4-1BBL with a T cell bispecific (TCB) antibody cotargeting tumor antigen (CEA or TYRP1) and CD3. In a 3D tissue culture of FAP+CEA+ colon adenocarcinoma with added human PBMCs, stimulation with FAP-4-1BBL and CEA-TCB led to a strong increase in IFNγ production by infiltrating T cells. Similar results were observed in 3D tissue cultures of lung adenocarcinoma, adenosquamous carcinoma, or melanoma without added human PBMCs.
Because CEA-TCB treatment alone induces 4-1BB upregulation on tumor-infiltrating T cells, the addition of FAP-4-1BBL is a rational combination. In human stem cell-engrafted NSG mice bearing xenograft gastric MKN45 tumors and murine NIH/3T3 fibroblasts, combination CEA-TCB plus FAP-4-1BBL increased CD8+ T cell infiltration, increased the CD8+/CD4+ T cell ratio (though not the CD8+/Treg ratio), and inhibited tumor growth without inducing immune cell accumulation in the liver. This model, however, may not have represented full immunocompetence, so in an immunocompetent tumor-bearing mouse model, the researchers showed that a surrogate murine FAP-4-1BBL synergized with a surrogate CEA-TCB, as well as with murine anti-PD-1, mediating tumor remission without off-tumor accumulation of immune cells in the liver.
With promising evidence that FAP-4-1BBL could pair well with T cell bispecifics or immune checkpoint blockades, Claus, Ferrara, and Xu et al. designed a similar fusion protein, CD19-4-1BBL, for targeting B cell lymphoma. In a culture of CD3-stimulated human PBMCs, CD19-4-1BBL increased production of IFNγ. In combination with a CD20-TCB, which alone upregulates 4-1BB on T cells, IFNγ was further upregulated.
Looking more deeply into the mechanism, the researchers used confocal fluorescence microscopy to demonstrate dynamic localization of CD19-4-1BBL during CD20-TCB cross-linking at sites of interaction between tumor and immune cells. This suggested that CD20-TCB cross-linking initiates an interaction between T cells and target cells, and that CD19-4-1BBL proteins are then redistributed to the synapse and provide costimulation. In mice bearing an aggressive form of lymphoma, CD20-TCB monotherapy showed some efficacy, but the addition of CD19-4-1BBL enhanced CD8+ T cell infiltration, increased the CD8+/CD4+ T cell ratio, increased granzyme B production, induced an earlier onset of antitumor effects, and completely eradicated tumors without signs of toxicity. Even in a lymphoma model with much lower CD20 expression, combination treatment resulted in improved tumor control correlating with accumulation of T cells in the tumor.
While monoclonal antibodies targeting 4-1BB have shown limited efficacy, 4-1BB has been successfully exploited to enhance the activity and persistence of third generation CAR T cells. Here, Claus, Ferrara, and Xu et al. show that an Fc-independent fusion protein cotargeting 4-1BBL and a tumor/stroma antigen in combination with tumor antigen-targeted T cell bispecific antibodies may be able to mimic the efficacy of CAR T cells using only off-the-shelf products.
by Lauren Hitchings
Meet the Researcher
This week, first co-author Christina Claus answers our questions.

What prompted you to tackle this research question?
We have at Roche a great and creative team of antibody engineering experts. In the last years, several engineered antibodies have been developed, like obinutuzumab or the T cell bispecific antibodies CEA-TCB or CD20-TCB, to help tumor-killing immune cells recognize the tumor cells.
Given the immunosuppressive environment of the tumor, we knew that pure redirection of the tumor-killing immune cells would not lead to complete tumor remission in most cases. Therefore, in December 2010, our lead scientists from the Roche Innovation Center Zurich (Pablo Umaña, Christian Klein, Marina Bacac) initiated the development of several immunomodulatory antibodies and fusion proteins, like tumor-targeted 4-1BB agonists, aiming to boost these tumor-killing cells to survive and to overcome the immunosuppressive tumor environment by providing supportive co-stimulation.
We were aware of clinical challenges conventional antibody based 4-1BB agonists faced at this time and we thought that a clever molecule design could help overcome these issues.
Our approach is to stay flexible in adapting and improving therapy strategies, as cancer is a very diverse and individual disease. The published tumor-targeted 4-1BB agonists reflect this strategy and are supposed to be combined with different cancer therapies to improve the life and therapy of patients with different tumor diseases.
What was the most surprising finding of this study for you?
I joined Roche Innovation Center Zurich in May 2011 fresh from academia. Before I had worked in basic immunology research where we always used “normal” antibodies to activate or block receptors to study immunological response, while not really focusing on the crosslinking of these antibodies.
At the Roche Innovation Center Zurich I joined a really great and supportive team who encouraged me with their creativity and positive spirit. I spent my first three years working on the development of assays and screening hundreds of molecules. Especially in the beginning this was quite repetitive, as negative results were frequent. However, my colleagues in antibody engineering kept me excited with their creativity and engagement in molecule design and their experience. To get the opportunity to follow the development of a drug from the laboratory bench to clinic was very often surprising. Suddenly, you become aware what pharmaceutical development really means. There is so much work and support in the background I was not aware of. Sharing and learning from so many diverse and experienced people has kept my work extremely interesting and engaging. I have realized that team spirit and diversity are as important as the love for science.
What was the coolest thing you’ve learned (about) recently outside of the lab?
The field of tumor immunology is booming. It is a really exciting time. There are so many new technologies like single cell sequencing and high throughput screening leading to an increasing amount of data and publications. This can be fascinating as well as overwhelming. Currently I am really interested in literature on tumor-specific T cell characterization. I mainly focus on the expression pattern of different surface molecules, but also their origin and the T cell receptor repertoire. Hopefully this will help to identify patients in different treatment groups and to identify the best therapy for each of these patients.
For more background on this study
We have partnered with ReFigure.org, a novel platform for collecting scientific figures. See this collection of figures describing the principles behind the approach followed in this study.



