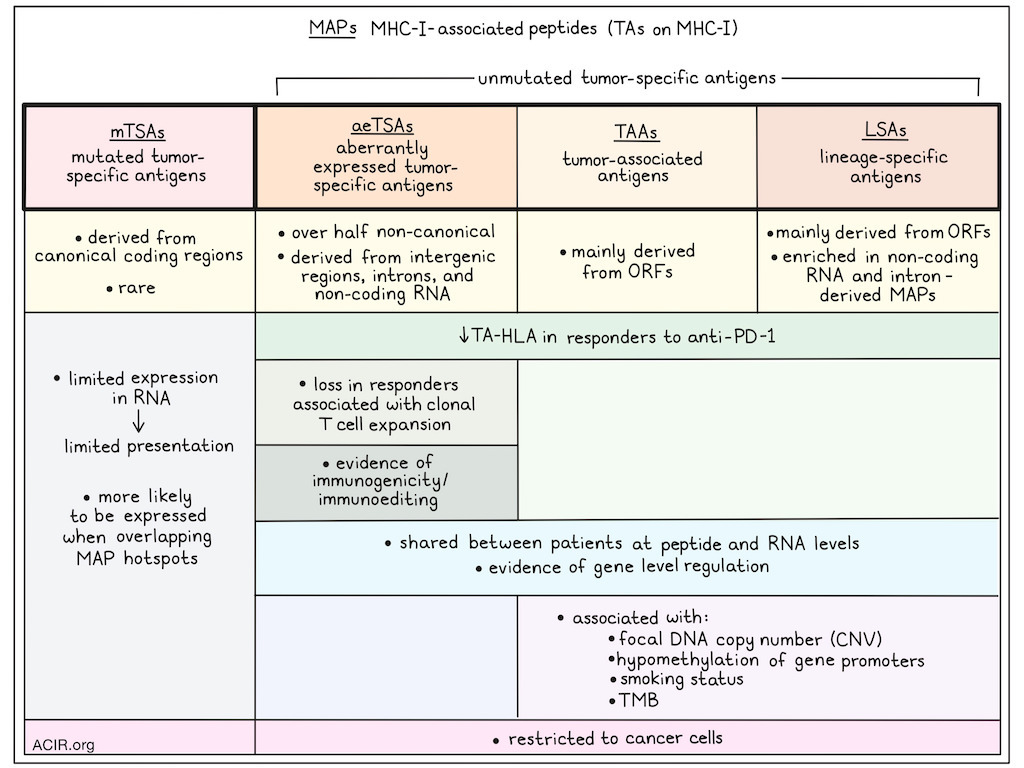
Recent discoveries of unmutated aberrantly expressed tumor-specific antigens (aeTSAs) derived from non-coding regions and embryonic transcriptional programs has garnered attention as targets for immunotherapy. Apavaloaei et al. explored cell surface presentation of peptide:MHCs derived from mutated and unmutated tumor antigens (TAs) in melanoma and non-small cell lung cancer (NSCLC), cancer types with high tumor mutational burden (TMB), and published their findings on new therapeutic targets in Nature Cancer.
The researchers analyzed immunopeptidomes (the sum of MHC-I-associated peptides [MAPs]) from 26 NSCLC, 12 cutaneous melanoma biopsies, and multiple melanoma cell lines with proteogenomics, and reanalyzed RNAseq and paired immunopeptidomics data from seven patient-derived melanoma cell lines. Bulk RNAseq was used to construct sample-specific databases, and tandem mass spectra (MS/MS) of MAPs from each sample were searched against these databases to identify signature peptide degradation patterns. This identified unique MAP sequences from which the RNA coding sequences, genomic locations, and expression in healthy and cancer tissue were assessed to determine the class of TA candidates. These MAP classes included: mutated tumor-specific antigens (mTSAs) from mutations in canonical or non-canonical regions not expressed in healthy tissue; aeTSAs based on absence or minimal expression in normal tissues, except the testis; lineage-specific antigens (LSA) when antigen expression was restricted to normal lung or skin; and tumor-associated antigens (TAAs) with significant expression in other healthy tissues and a minimal two-fold overexpression in TCGA.
This approach retrieved 90 TAs each from NSCLC and melanoma, and 434 TAs from the melanoma cell lines. Over half of the detected aeTSAs were non-canonical, derived largely from intergenic regions, introns, and non-coding RNAs. TAAs and LSAs were mainly derived from open reading frames (ORFs), and LSAs in melanoma were also enriched in non-coding RNA and intron-derived MAPs. mTSAs were from canonical coding regions, but very rare in both cancer types.
To explore the reason for this limited number of mTSAs, the researchers found that most exonic non-synonymous mutations can code for a MAP with adequate predicted MHC-I-binding affinity. Expanding the search database to include not just single-nucleotide variants, but also multi-nucleotide variants limited additional predicted mTSAs. The retrieved candidates were further assessed using transcriptomic analysis of tumor and healthy tissues to determine which were genuine mTSAs, and most were excluded due to high expression in healthy tissue. Further, maximally, only 32% of the exome-derived mutations were expressed in the RNAseq reads from melanoma cell lines, suggesting that most exonic mutations do not generate MAPs, as this requires RNA expression. Overall, RNA expression levels for predicted mTSAs that were undetected by MS were significantly lower than for unmutated TAs, suggesting transcripts coding for predicted mTSAs were less processed for antigen presentation, mainly due to low RNA expression.
The researchers previously showed that MAPs preferentially derive from MAP hotspots, which are actively transcribed and translated, resulting in a high proportion of defective ribosomal products. Of all mutations, 25% of those in NSCLC and 33% of those in melanoma overlapped with MAP hotspots. Predicted mTSAs derived from mutations outside these hotspots were less likely to generate MAPs than those in the hotspots in both cancer types.
To determine whether a higher MS sensitivity could increase the detection of predicted mTSAs, 21 predicted mTSAs from NSCLC were selected for targeted MS analysis, which is more sensitive, but can only be used for a limited number of MAPs. Only one predicted mTSA was detected by targeted MS, which was an mTSA not expressed in healthy tissues. Therefore, the lack of MS detection of predicted mTSAs may not be due to the detection threshold of MS analyses and instead may be due to being outside of MAP hotspots.
The researchers then focused on determining how the classes of unmutated TAs (aeTSAs, LSAs, and TAAs) play a role in antitumor immunity, using pre-treatment biopsies from patients with NSCLC and melanoma treated with ICB. TA presentation was inferred based on the expression of both the TA and the predicted cognate HLA, identifying TA–HLA pairs. In both NSCLC and melanoma datasets, high numbers of TA–HLA pairs were detected. However, no differences were detected in the number of TA–HLA pairs in pre-treatment samples between ICB responders and non-responders. Using a dataset from anti-PD-1 therapy in melanoma, responders were found to have a decrease in the number of unmutated TA–HLA pairs during therapy, which was not observed in non-responders. In particular, a loss of aeTSA–HLAs, but not LSAs or TAAs, in responders was correlated with the number of T cell clones expanding during therapy.
Using the functional expansion of specific T cells (FEST) assay, the researchers determined whether the lost aeTSAs in responders were immunogenic and could be recognized by CD8+ T cells. For this, T cells isolated from healthy donors were stimulated with autologous cells pulsed with synthetic aeTSAs, and TCR clonotypes responsive to those antigens were identified based on their expansion. Twelve melanoma aeTSAs were assessed, which were all immunogenic and induced polyclonal T cell expansion. Further, cytotoxicity assays with 6 HLA-A*02:01-binding aeTSAs showed that T cells primed against four of these antigens expanded and specifically killed aeTSA-expressing B lymphoblastoid cells. Finally, T cells primed against two of the aeTSAs were assessed for their ability to kill melanoma cells endogenously expressing these aeTSAs. The T cells showed cytotoxicity against the tumor cells, even though the aeTSA RNA expression on these cells was relatively low, suggesting these antigens are highly immunogenic.
Apavaloaei et al. found that unmutated TAs from melanoma and NSCLC were shared between patients at the peptide and RNA levels. TCGA multiomics data revealed a correlation between the RNA expression of TAs and their corresponding source genes, suggestive of gene-level regulation. In melanoma, TAAs and LSAs were associated with the focal DNA copy number (CNV) and the hypomethylation of source gene promoters. In lung squamous cell carcinoma, the CNV was the most important contributor to TAA expression, while LSAs were less common. In lung adenocarcinoma, TAA and LSA expression correlated with smoking status and TMB. Overall, these data suggest that TA expression may reflect cancer cell programs shared between patients.
Finally, published scRNAseq datasets from melanoma and NSCLC were used to validate that unmutated TA expression was associated with cancer cells only, and no other cells in the TME. This revealed that, like LSAs and TAAs, aeTSAs are restricted to cancer cells, suggesting these antigens are cancer-specific.
Overall, the data in this study suggest that unmutated TAs, and in particular aeTSAs, are immunogenic, cancer-specific, and shared between patients, making them appealing candidates for the development of novel vaccine or cellular immunotherapy approaches.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Anca Apavaloaei answered our questions.

What was the most surprising finding of this study for you?
Our lab has previously described the presence of numerous unmutated non-canonical HLA I-associated tumor antigens in cancers with low-to-medium mutational burden, such as AML, ovarian cancer, and breast cancer. In melanoma and NSCLC, we expected to find many mutated tumor antigens, which for a long time were believed to be the main therapeutic targets in these highly mutated cancers. Surprisingly, we found very few. Instead, non-canonical tumor antigens were much more abundant and shared, representing an important therapeutic opportunity across cancers, independent of mutational burden.
What is the outlook?
Our team is doing extensive validation studies to prioritize the non-canonical tumor antigens most promising for clinical trials, which we hope to launch within two years.
Who or what has been a major source of inspiration or motivation for you throughout your career?
The lead author, Dr. Claude Perreault, has been an exceptional scientific mentor throughout my MSc and PhD training in his lab. He continues to be a major source of inspiration as I navigate my scientific career.



