CD4+ T cells are known to “help” CD8+ T cells by licensing their cytotoxic functions in tumors, but exactly how this help is given is not entirely understood. To better understand how CD4+ T cells contribute to CD8+ T cell responses, Espinosa-Carrasco et al. developed a tumor and adoptive cell therapy (ACT) model in which they could study the relationship between these cells, and found that CD4+ T cells formed critical triads with CD8+ T cells and APCs in order to license CD8+ T cell-mediated cytotoxicity and antitumor immunity. Their results were recently published in Cancer Cell.
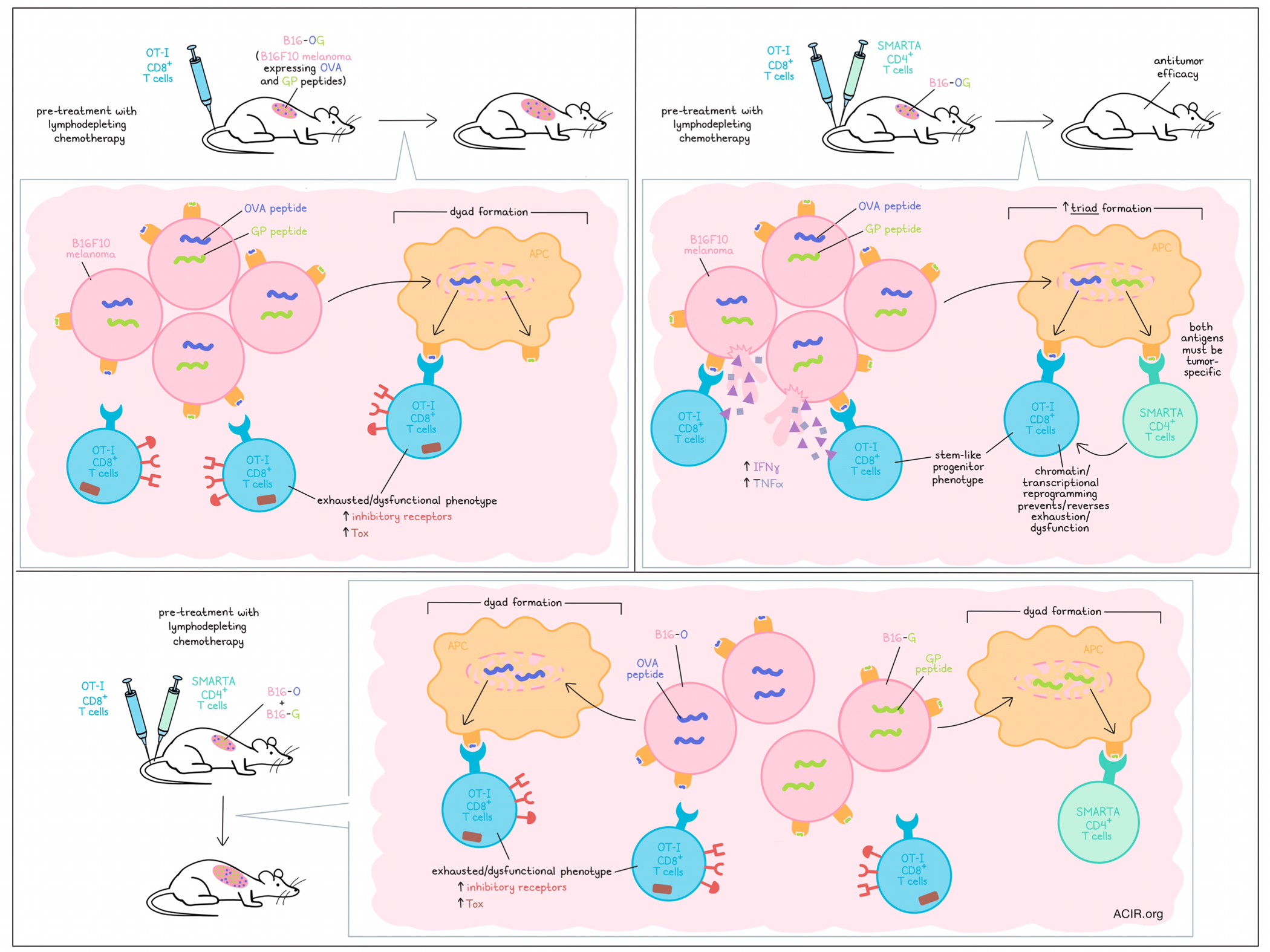
To begin, Espinosa-Carrasco et al. engineered the B16F10 melanoma model to express (1) the SIINFEKL peptide from the OVA antigen for recognition by OT-I CD8+ T cells, (2) the glycoprotein epitope GP61–80 from LCMV for recognition by SMARTA TCR-transgenic CD4+ T cells, and (3) a fluorescent tag. The resulting B16-OG cells grew aggressively upon implantation into mice, forming large tumors within two weeks. Mimicking clinical protocols for ACT, the researchers pre-treated mice with lymphodepleting chemotherapy, then transferred activated OT-I cells, SMARTA cells, or both. While transfer of either cell type alone allowed for continued tumor progression, the combination of CD8+ and CD4+ T cells resulted in complete tumor elimination and long-term tumor-free survival in 100% of mice. This effect was lost when OT-I cells were combined with CD4+ T cells specific for an irrelevant antigen, suggesting that both types of responses must be tumor antigen-specific for antitumor efficacy. Similar results were observed in an engineered MCA205-OG tumor model.
Looking within tumors, the researchers found that 8-9 days post-transfer, OT-I cell infiltration was similar, regardless of whether they were transferred with SMARTA cells. However, OT-I TILs that had been transferred alone produced few effector cytokines and expressed higher levels of inhibitory receptors and Tox, while those transferred in tandem with SMARTA cells produced much more IFNγ and TNFα and showed little to no expression of inhibitory receptors or Tox, suggesting that CD4+ T cells mediate a functional difference in CD8+ T cells. Further, this effect was observed in tumors, but not tdLNs, indicating that CD4+ T cell support is provided on-site in the TME.
To determine whether CD4+ T cells could reverse CD8+ T cell exhaustion, the researchers transferred OT-I cells into tumor-bearing mice, followed by transfer of SMARTA cells 10 days later. While tumors continued to progress with OT-I cells alone, the late addition of SMARTA cells rescued OT-I production of IFNγ and TNFα, and induced tumor regression. To better understand how this licensing occurred, the researchers performed RNAseq and found that the inclusion of SMARTA cells reduced the expression of various dysfunction-related transcription factors and inhibitory receptors, and instead induced expression of transcription factors and molecules associated with stem-like progenitor states. These results were also reflected in ATACseq data, which showed differentially accessible regions in exhaustion and stem/progenitor genes.
Evaluating the clinical relevance of these findings, the researchers compared profiles identified in mouse models with data from patients with metastatic melanoma lesions who received lymphodepleting chemotherapy followed by ACT. Here, the team identified a subset of CD39-CD69- stem-like TILs resembling OT-I cells that had been transferred with SMARTA cells. This subset in patients was associated with complete regression in responders, and was notably lacking in non-responders. In contrast, a CD39+CD69+ subset identified in non-responders resembled OT-I cells that were transferred without SMARTA cells in mice.
Next, the researchers investigated whether CD4+ T cells might be acting directly on tumor cells, but found that B16-OG tumor cells engineered to be MHC-II-deficient were eliminated with similar efficacy. However, when CD11c+ DCs were conditionally depleted in vivo, the antitumor effect of OT-I + SMARTA cells was lost, suggesting that APCs have a role in CD8+ T cell licensing. To separate out the presentation of the different tumor antigens by APCs, the researchers generated B16-O and B16-G tumor cell lines, each expressing only one of the respective model antigens, and then co-transferred both tumor cell lines into mice to generate heterogeneous B16 O+G tumors. Unlike the homogeneous B16-OG tumors B16 O+G tumors failed to respond to treatment with a combination of OT-I and SMARTA cells, despite similar antigen expression levels and similar tumor infiltration. In the B16 O+G tumors model, the OT-I cells co-transferred with SMARTA cells more closely resembled OT-I cells that had been transferred alone in the B16-OG model, expressing an exhausted/dysfunctional phenotype.
Confocal microscopic analysis of established B16 O+G tumors showed that there was little overlap between tumor regions expressing OVA versus GP antigens, suggesting that antigen-presenting cells in a given region would likely only pick up one antigen at a time, rather than both, as they would in the B16-OG model. Investigating whether co-presentation of antigens was the key to antitumor efficacy in the B16-OG model, the researchers developed a spatial analysis model in which cancer cells, OT-I cells, SMARTA cells, and APCs were each fluorescently tagged with unique markers. This revealed that in the B16-OG model, 3.5-fold more three-cell clusters, or “triads” of OT-I cells, SMARTA cells, and APCs formed than in the B16 O+G model. Further, when B16-OG cancer cells were injected into one flank while B16-O cancer cells were injected the opposite flank of the same mouse, only the B16-OG were eliminated by OT-I and SMARTA combination therapy, highlighting the importance of co-expression of tumor antigens on the same APCs within tumors to support CD8+ T cell-mediated antitumor immunity. Triad formation was also found to be relevant in responses to checkpoint blockade, as B16-OG, but not B16 O+G tumors were rejected upon treatment with anti-PD-1/anti-PD-L1.
Again turning to patient data, the researchers assessed the spatial orientation of CD4+ T cells (non-Tregs), CD8+ T cells, and APCs in patients with malignant plural mesothelioma who were treated with checkpoint blockade. While dyads of CD4+ T cells and APCs were not associated with responses, triad formation could delineate responders from non-responders. Further, PD-1+CD8+ T cells that were part of triads expressed higher levels of IFNγ and granzyme B compared to those not in triads.
Overall, these results suggest that the formation of triads of CD4+ T cells, CD8+ T cells, and APCs, in which APCs co-presenti relevant tumor antigens to each T cell type, allows for CD4+ T cell-mediated licensing and reprogramming of CD8+ T cells towards a more progenitor-like state, preventing and even reversing exhaustion and dysfunction. The formation of these triads was further associated with antitumor responses and clinical outcomes in mice and patients. Triad formation could therefore serve as both a biomarker for response and a target for improving various forms of cancer immunotherapy.
Write-up and image by Lauren Hitchings



