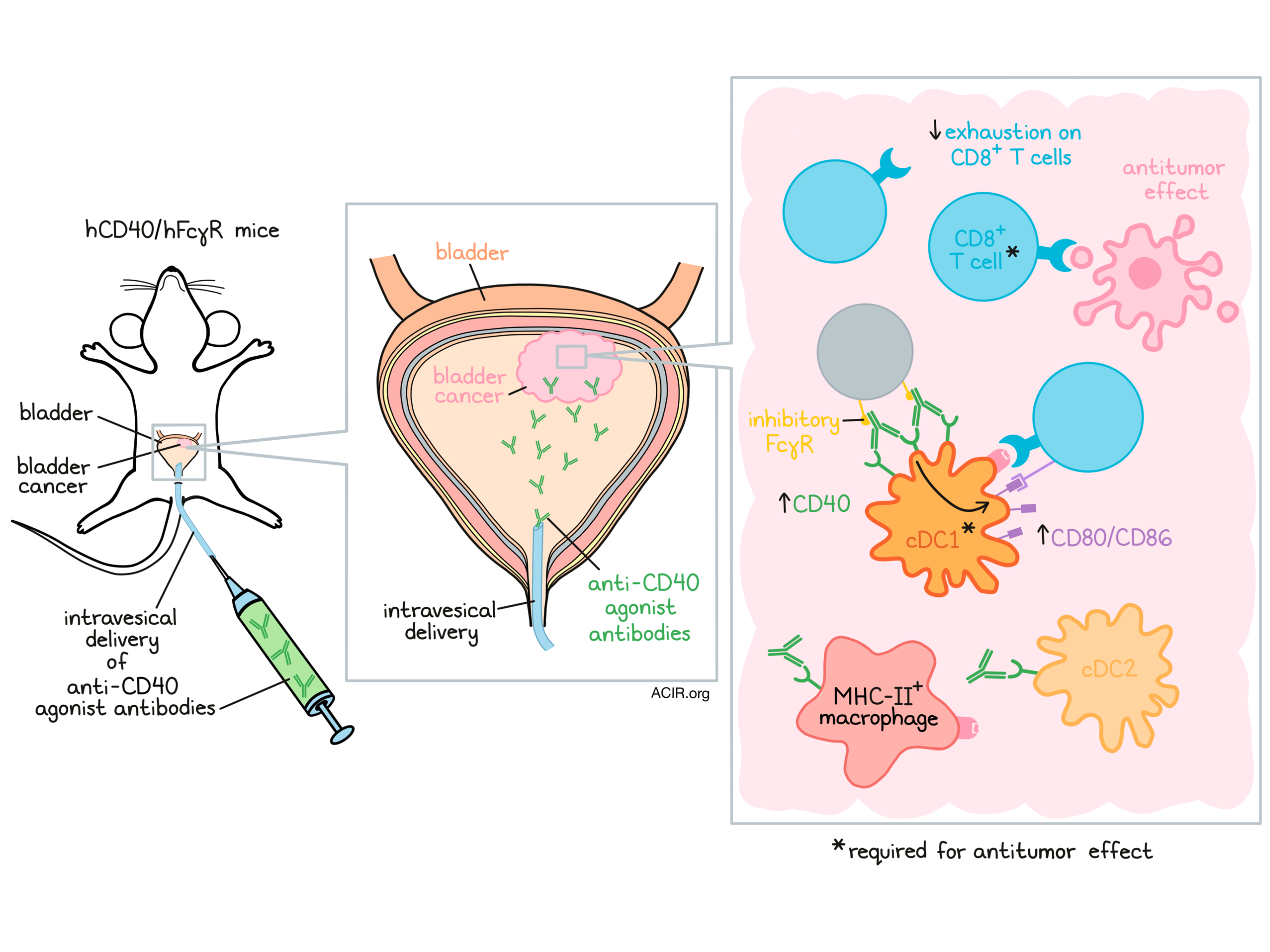
When patients are diagnosed with non-muscle-invasive bladder cancer, intermediate- to high-risk patients typically undergo transurethral tumor resection, followed by intravesical treatment with Bacille Calmette-Guérin (BCG) as the standard of care. However, up to 75% of patients ultimately become unresponsive to this treatment. Even with the recent approval of anti-PD-1 therapy in the BCG-unresponsive setting, current treatment options are insufficient at preventing relapse. In a recent study, Garris and Wong et al. explored the possibility of using an anti-CD40 agonist antibody to treat bladder cancer; their results were recently published in Science Translational Medicine.
Garris and Wong et al. began by using an orthotopic bladder implantation model in which tumors initially invaded the bladder tissue, but not the muscle. Within these tumors, immune infiltration was increased compared to healthy bladder tissue. Further, expression of CD40 was increased within tumors, and was expressed mainly by dendritic cells (DCs), particularly conventional type 1 DCs (cDC1).
Next, the researchers treated tumor-bearing mice with intravesical delivery of the CD40 agonist antibody 1C10, which stimulates CD40 through cross-linking, enabled by Fc binding to the murine inhibitory FcγRIIb receptor, and saw reduced tumor burdens in the bladder compared to both BCG and control treatments. Within the immune microenvironment, CD8+ T cell exhaustion signatures, including expression of PD-1 and Lag3, were reduced. CD8+ T cell depletion demonstrated that the antitumor effect of 1C10 was CD8+ T cell-dependent.
To determine the role of DCs in the antitumor response, the researchers turned to Batf3-/- mice, which lack cDC1s. In this setting, implanted bladder tumors grew more quickly than in wild-type mice, with some tumors even invading the muscle. Similarly, Batf3-/- mice that were exposed to the carcinogen BBN through drinking water developed higher tumor burdens in their bladders than wild-type mice. Together, these results suggested that cDC1s likely play a role in immune surveillance of bladder tumors, even before treatment.
When bladder tumor-bearing Batf3-/- mice were treated with anti-CD40 agonist antibody, the antitumor effect was lost and CD8+ T cell infiltration was reduced compared to wild-type mice. Further, even subcutaneous tumors, which are typically highly responsive to 1C10, were refractory to treatment in Batf3-/- mice, suggesting that Batf3+ cDC1s are required for early tumor control and responses to anti-CD40. Further supporting this, fluorescent labeling in wild-type tumor-bearing mice showed that intravesical anti-CD40 bound mainly to cCD1, cDC2, and MHC-II+ macrophages.
In an effort to make their mouse studies more clinically relevant, Garris and Wong et al. crossed humanized FcγR mice (expressing all human FcγRs and no mouse FcγRs) with mice expressing human CD40; the result was hCD40/hFγR mice that were humanized for both CD40 and FcγRs. Using this model, the researchers evaluated a fully human anti-CD40 agonist antibody CD870,893, which has previously been tested in clinical trials, and compared it to 2141-V11, which has the same CD40 binding Fab as CD870,893, but was Fc-optimized to bind FcγRIIb more strongly. In mice bearing subcutaneous MB49, UPPL1541, or BBN963 bladder tumors, 2141-V11 induced nearly 100% complete responses, which were associated with a substantial increase the DC-associated T cell costimulatory factors CD80 and CD86. In mice bearing bilateral subcutaneous tumors, 2141-V11 induced potent primary tumor regression and an abscopal effect, suggestive of systemic antitumor immunity.
While BCG therapy induced some antitumor effect in mice bearing subcutaneous tumors, it did not induce any durable tumor remissions. Testing 2141-V11 in BCG-unresponsive tumors showed that 2141-V11 induced and sustained both local and systemic antitumor activity, suggesting that BCG does not impair future anti-CD40 agonist antibody treatment, and that anti-CD40 agonists can potentially rescue responses in BCG-refractory tumors.
Moving towards intravesical delivery, the researchers first performed dose titration studies of 2141-V11 in non-tumor-bearing mice, and while intraperitoneal delivery of 2141-V11 showed dose-limiting systemic effects, the same was not observed with intravesical delivery, likely due to the relative impermeability of the bladder walls to large molecules, like antibodies. When the same dose titration studies were done in tumor-bearing mice, there was a degree of platelet reduction and serum transaminase elevation, which may have been driven by systemic absorption associated with advanced, highly vascular tumors. Interestingly, there was no clear relationship between dose and efficacy, with the lowest dose actually showing both the highest efficacy and the lowest toxicity. These results suggest that dosing should be carefully considered, and that the maximum tolerated dose may not inherently be the most effective.
Testing the efficacy of intravesical delivery of 2141-V11, the researchers observed potent antitumor effects and improved survival in mice bearing both advanced (day 6) and early (day 3) MB49 bladder tumors. After 60 days, surviving mice rejected a rechallenge of tumors at 10x the initial tumor dose, suggestive of long-term antitumor immunity. These results were confirmed in mice bearing UPPL1541 bladder tumors, which closely recapitulate the luminal molecular subtype of human high-grade urothelial cancer. Like in the subcutaneous models, 2141-V11 was also effective against bladder tumors that were refractory to BCG. Overall, the researchers observed 50-60% complete responses in orthotopic models. The researchers hypothesized that this lower efficacy relative to the results in subcutaneous tumors could be due to differential access to the tumor-draining lymph nodes and limited accumulation of 2141-V11.
Given the potent antitumor effects of Fc-optimized anti-CD40 agonist antibody 2141-V11 in mice humanized for both CD40 and FcγRs, and the reduced toxicity with intravesical delivery, it is possible that this approach could have a similar benefit in a clinical setting, including in patients who are refractory to standard-of-care intravesical BCG therapy, warranting clinical studies.
By Lauren Hitchings
Meet the researcher
This week, we interviewed lead author David Knorr.

What prompted you to tackle this research question?
For years, immunologists have used CD40 antibodies to help adjuvant the immune response in several model systems, including cancer. Thus, there was initially great interest in developing agonist anti-CD40 antibodies into a novel therapeutic for patients with various malignancies. Several antibodies were tested in clinical studies throughout the 2000s, however, many of them failed to elicit antitumor activity at their maximum tolerated dose (limited by thrombocytopenia and transaminitis). During this time, our group described the unique requirement for agonist antibodies to engage the inhibitory Fc gamma receptor, FcγRIIB. While antibodies are traditionally envisioned to elicit ADCC through engagement of activating FcγRs, the in vivo requirements for optimal anti-CD40 agonist antibody therapy rely on binding FcγRIIB. Because binding to FcγRIIB does not lead to cellular activation, it is thought to serve as a scaffold, allowing for optimal receptor clustering. This clustering is important when considering the natural biology of TNF-superfamily members like CD40, which require trimerization for downstream signaling.
Understanding the requirement for FcγRIIB, we then engineered the Fc domain of a fully human anti-CD40 antibody that had previously been tested in humans. Compared to its parental IgG2 counterpart, Fc-engineering of an IgG1 anti-CD40 antibody for FcγRIIB engagement led to enhanced T cell activation, as well as improved activity in several tumor models. However, this also led to enhanced toxicity (thrombocytopenia and transaminitis, as in patients) when given to humanized mice (expressing hCD40 and hFcγRs). Understanding that CD40 engagement could locally activate antigen-presenting cells, we then tested an in situ vaccination approach which led to robust local and systemic (abscopal-like) responses. This intratumoral approach is now being tested in patients with injectable solid tumors as a phase I clinical study (NCT04059588).
Having demonstrated that local delivery of our Fc-enhanced agonist CD40 antibody could lead to potent local and systemic immunity, we looked for other clinical scenarios where local immunotherapy has been successful. We were initially attracted to patients with non-muscle invasive bladder cancer (NMIBC), where the use of intravesical BCG has been standard of care for over 5 decades. The relative impermeability of the bladder urothelium provides an exceptional opportunity to test strong immune agonist treatments while minimizing systemic toxicity. With the tremendous insight and support from our colleagues at MSKCC and the Bladder SPORE program, we were up and running! Our pre-clinical studies found that intravesical treatment with CD40 agonistic antibodies can provide both local and systemic immunity against tumors, without evidence of systemic toxicity. We now hope to investigate this biology in humans as part of our phase I clinical study opening for NMBIC this summer.
What was the most surprising finding of this study for you?
I suppose what was most surprising to us was the level of systemic antitumor immunity elicited by intravesical CD40 agonist antibodies. We knew before that CD40 agonist antibodies could drive abscopal effects, but in prior studies this was tested in a similar tissue compartment such as skin to skin. It was fascinating to see that a bladder-driven immune response could be protective when mice were rechallenged with tumors in the skin, and suggests that CD40 agonist driven immunity provides long-term systemic antitumor immunity. This is critically important in patients with high-risk NMIBC, because those failing therapy and progressing to muscle-invasive disease are at risk for dissemination to draining lymph nodes and systemic organs, at which point the disease is metastatic.
What was the coolest thing you’ve learned (about) recently outside of work?
While many of our normal activities were put on hold much of the last year, it was great to see the resiliency of our hospitals with our city being at the center of the initial pandemic. That same energy and positive outlook remains as things get back to normal. We are moving to a nearly fully vaccinated and mask-free campus, and are getting to enjoy all of the things we love to do outside of work, including local arts, food, and culture (and for both Chris and Jeff being new fathers!).



