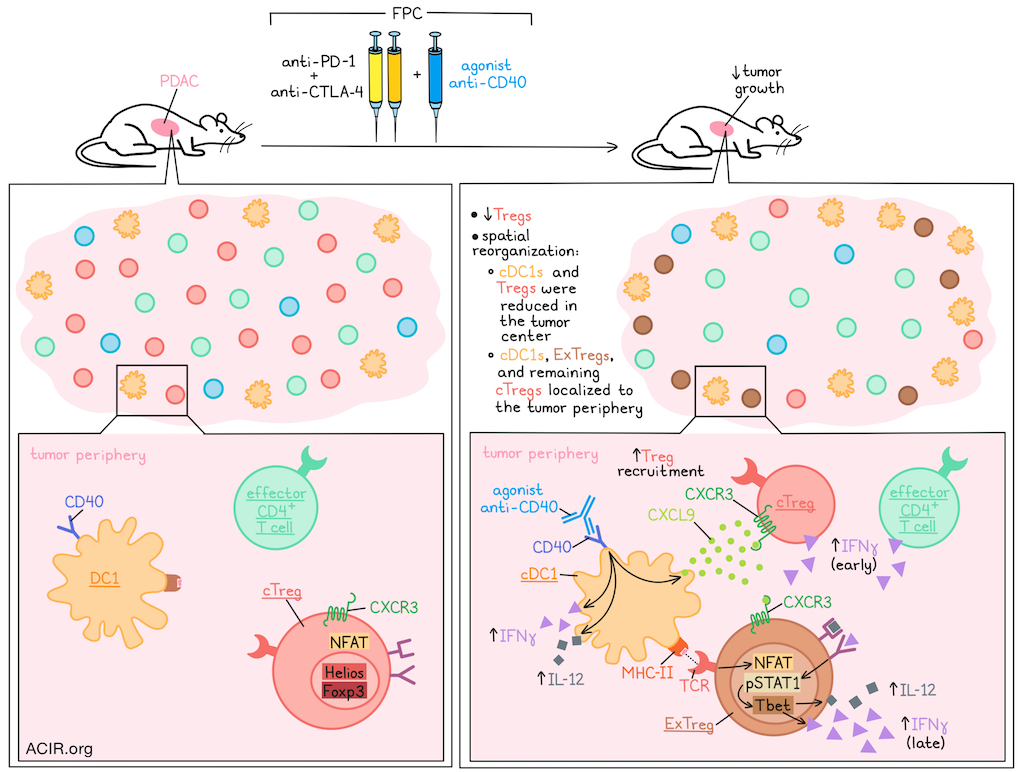
Recent investigations into the use of agonist anti-CD40 for cancer immunotherapy have shown that this treatment not only enhances APC functions, but also reduces regulatory T cells (Tregs) within the tumor microenvironment (TME), despite a lack of CD40 on these cells. Exploring the mechanism by which this occurs, and whether it might contribute to antitumor efficacy, Maltez et al. found that agonist anti-CD40 induced spatial reorganization in the TME and, via activated cDC1s, converted conventional Tregs (cTregs) into “ExTregs” with Th1-like functions. Their results were recently published in Immunity.
To begin, Maltez et al. established that in a PDAC tumor model treated with dual ICB (anti-PD-1 plus anti-CTLA-4; PC), the addition of agonist anti-CD40 (F) resulted in tumor control and a loss of Tregs, dependent on host CD40 expression. After FPC treatment, remaining Tregs localized to the tumor periphery, despite more homogeneous distribution of effector CD4+ and CD8+ T cells throughout. Similar results were also observed in other tumor models.
Given that cDC1s are a major target of CD40 agonism and were the dominant CD40+ population in their PDAC model, the researchers investigated their role in Treg reduction using Batf3-/- mice. This showed that cDC1s were required for the spatial reorganization and reduction of Tregs after FPC. Further investigation using IL-12p40-/- mice, IFNγ-/- mice, and cytokine blockades showed that both IL-12 and IFNγ, typically produced by activated cDC1s, were also required.
Next, the researchers investigated whether Treg reduction occurred through apoptosis, efflux, or phenotypic conversion. There was not enough evidence of apoptosis or efflux to account for the observed Treg loss, so the researchers focused their attention on Treg conversion through loss of Foxp3. To study this, they developed a lineage-tracing mouse model in which Foxp3+ Tregs were permanently tagged prior to treatment with FPC. After treatment, tagged cells were evaluated for Foxp3 expression, which showed that a substantial portion of former conventional Tregs (cTregs) had converted in the tumor to become “ExTregs” through loss of Foxp3 expression. This effect was specific to the tumor site, and similar results were observed in other tumor models. Consistent with this local conversion, blockade of lymph node egress using FT720 yielded similar proportions of cTregs and ExTregs. These effect of this phenotypic conversion also lasted through day 10 after FPC treatment, with no recovery of cTregs, suggesting the change was likely permanent.
Looking at transcription factors involved in the conversion of Tregs from cTregs to ExTregs, the researchers found that expression of Helios, which controls Foxp3 expression and immunosuppressive functions, was reduced in Tregs after FPC treatment, while expression of Tbet, which largely controls Th1 effector cell responses (including IL-12 and IFNγ), was upregulated in ExTregs, coinciding with the loss of Foxp3. Further, pSTAT1, indicative of IFN signaling and a precursor to Tbet expression (and subsequent IFNγ and IL-12R upregulation), was increased in ExTregs in treated mice, but was lost in settings of IL-12 or IFNγ blockade.
Next, Maltez et al. examined the functions of ExTregs in situ, and found that a large portion of ExTregs upregulated Ifng transcripts after FPC treatment (but not ICB alone), as did a modest portion of remaining cTregs. Blockade of IL-12 or IFNγ abrogated the increase in Ifng transcripts within both cTregs and ExTregs.
Investigating the original source of IFNγ in this apparent feedforward mechanism, in which IFN signaling induces ExTreg development and further IFNγ production, the researchers found that early after agonist anti-CD40 treatment, CD4+ effector T cells and cTregs were the dominant sources of IFNγ, but by 24-48 hours, ExTregs became the dominant source.
Turning their attention towards the spatial reorganization of Tregs observed after FPC treatment, the researchers hypothesized that the IFNγ-inducible Cxcl9–Cxcr3 chemokine axis might contribute. They found that Cxcl9+ cells increased after treatment, concurrent with the increase in Ifng+ cells, and became physically closer to Ifng+ cells over 48 hours. The density of Cxcl9+ cells and Ifng+ cells also increased in the tumor periphery compared with the center. Further investigation revealed that after agonist anti-CD40, Cxcl9+ cDC1s were increased in the tumor periphery and became closer to both cTregs and ExTregs, which more frequently expressed Cxcr3 than non-Tregs. By 48 hours after treatment, most Treg-lineage cells in the tumor periphery were ExTregs and expressed Cxcr3.
To determine whether there were direct pMHC-II–TCR interactions between cDC1s and Tregs colocalizing in the tumor periphery, the researchers treated wild-type mice with an MHC-II-blocking antibody prior to FPC treatment. This abrogated the loss of cTregs, the residual localization of Tregs to the tumor periphery, and the increased proximity between Tregs and cDC1s. As further evidence of pMHC-II–TCR engagement, the researchers observed increased nuclear translocation of NFAT1 (a marker of recent TCR engagement) in both cTregs, ExTregs, and effector CD4+ T cells (particularly in the tumor periphery and in proximity to cDC1s), starting shortly after agonist anti-CD40 treatment, and further increasing over time. This effect was also abrogated by MHC-II blockade.
Finally, Maltez et al. and colleagues evaluated samples from their recent stage Ib/II clinical trial investigating neoadjuvant treatment with agonist anti-αCD40 and found that tumors showed evidence of an increased IFNγ signature, enrichment of APCs (including DCs), and increased Th1-like CD4+ T cells in the TME compared to baseline or control tumors. Evaluating specimens from both tumor centers and tumor borders, the researchers found that in treated patients, MHC-II+ DCs were enriched at the tumor border versus the center. The proportion of Tbet+Foxp3+ T cells was also increased in tumors from patients treated with anti-CD40, and these cells were concentrated at the tumor borders. Finally, the researchers stratified patients by longer or shorter disease-free survival (DFS), and found that patients with longer DFS had a trend toward lower densities of Foxp3+ T cells in the TME compared to patients with shorter DFS.
Overall, these results suggest that agonist anti-CD40 acts primarily on cDC1s, inducing Cxcl9 that preferentially recruits Cxcl3+ Tregs to the tumor periphery. There, Tregs are exposed to increased IL-12 and IFNγ (initially derived from activated cDC1s, CD4+ effector T cells and cTregs), inducing pSTAT1 signaling and expression of Tbet. Tregs also directly engage with cDC1s via MHC-II–TCR interactions, inducing NFAT1 nuclear translocation and cellular activation. These events cumulatively result in loss of Helios and Foxp3 expression, converting cTregs into ExTregs with a Th1-like phenotype and high IFNγ expression that could contribute to antitumor immunity, and could be further harnessed for cancer immunotherapy.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Vivien Maltez answered our questions.

What was the most surprising finding of this study for you?
One of the most surprising findings was that agonistic anti-CD40 therapy, not CTLA-4 blockade, was driving an acute Treg effect. CTLA-4 has long been associated with Treg biology, so we initially expected it to play the dominant role. Instead, CD40 emerged as the critical trigger, acting through cDC1s to reshape Treg fate. That result came relatively early, and immediately suggested we were seeing something fundamentally different in how these therapies operate. It was an exciting moment, but it also set us on a longer path to fully define the mechanism behind this unexpected axis.
What is the outlook?
A major next step is resolving the functional diversity of the Treg states we observed after therapy. We identified two populations: “reprogrammed” Tregs that retained Foxp3 but acquired effector features, and fully converted ExTregs that lost Foxp3 altogether. For translation, this distinction is critical: do we need complete destabilization, or is partial reprogramming sufficient to support antitumor immunity? Defining these thresholds will shape how we design therapies to safely harness Treg plasticity. More broadly, this work reframes Tregs not as fixed suppressors, but as dynamic cells that can be redirected in cancer.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
I would remind myself that while intelligence matters, it’s persistence that ultimately carries you through a scientific career. Early on, it’s easy to focus on who seems the “smartest”, but progress is really driven by curiosity, resilience, and the willingness to stay with hard problems over time. Experiments fail, ideas evolve, and momentum can be slow – but that’s the process. The ability to keep going, refine your thinking, and stay excited about the question is what makes the difference in the long run.



