Notice: The article we intended to feature this week was briefly delayed in publication. We will share it with you as soon as the embargo is lifted.
In the meantime, we encourage you to revisit last week's feature — it's well worth a read. If you've already read it, skip to the bottom of the page for our brand new spotlighted articles.
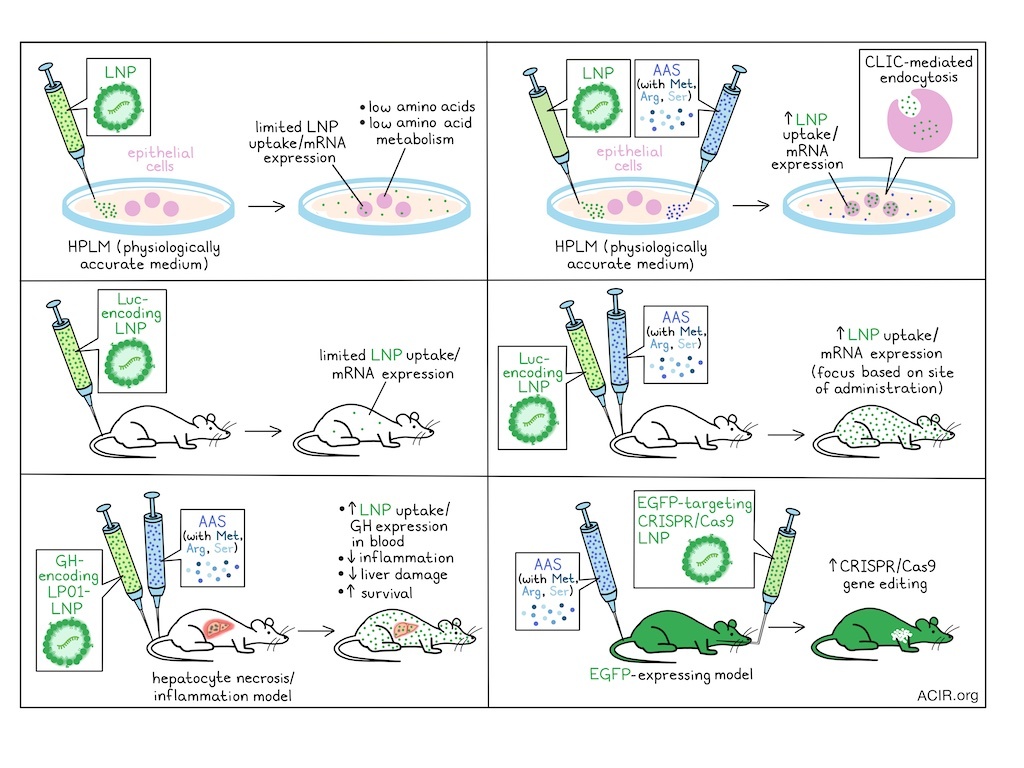
Lipid nanoparticles (LNPs) have allowed for the delivery of therapeutic mRNA for various therapeutic applications. However, in vivo delivery efficacy is often limited, and research is focused on optimization of LNP formulations. In recent work published in Science Translational Medicine, Chen, Wang, et al. assessed how the in vivo metabolic state impacts LNP delivery, and whether this can be modulated to improve delivery efficacy.
The researchers started by assessing cells cultured in either conventional medium (RPMI 1640 supplemented with fetal bovine serum) or human plasma-like medium (HPLM), which better simulates the physiologic metabolome. Compared to RPMI, HPLM contains lower concentrations of amino acids and glucose, similar levels of salt, and additional metabolites present in human plasma, such as lactate and urea. Lung- or colon tissue-derived epithelial cell lines were cultured for 7 days in these media, and treated with LP01 lipid-based LNPs with an enhanced green fluorescent protein (EGFP) reporter. This revealed a 50-80% reduction in EGFP expression when cells were cultured in the more physiologically accurate HPLM.
Based on these results, the researchers investigated the roles of specific metabolites and pathways in regulating LNP uptake through bulk metabolomic and transcriptomic analyses of cells cultured in these media. Analysis of metabolite profiles revealed that multiple amino acids, particularly methionine and arginine, were lower in cells cultured in HPLM. Pathway enrichment analysis showed that the most strongly enriched pathways were related to amino acid metabolism. Bulk RNAseq also revealed downregulation of amino acid metabolism pathways in cells cultured in HPLM conditions.
These data led the researchers to hypothesize that disruptions in amino acid metabolic pathways may contribute to impaired LNP uptake under physiologic conditions. HPLM was supplemented with various individual amino acids to levels found in RPMI, and cell lines were cultured in the different media and subjected to LNP delivery of EGFP. Supplementation with methionine, arginine, and serine enhanced EGFP expression by more than 2-fold. Based on these findings, Chen, Wang, et al. inferred that co-administration of LNPs with amino acid supplementation (AAS) may enhance the LNP delivery in physiologic environments in vivo.
To determine appropriate amino acid supplementation concentrations, methionine, arginine, and serine were assessed relative to their normalized levels in RPMI (X) – first individually, and then in combination. The optimal concentrations for formulating the AAS were identified as 30X methionine, 10X arginine, and 30X serine. To assess the effects of AAS on LNP delivery, cells were exposed to varying LNP concentrations and two lipid formulations, with or without AAS. The presence of AAS increased luminescence intensity by 5- to 10-fold. Further, different cell lines derived from several tissues also showed this enhancement, suggesting it can be broadly applied across various cell types.
To understand how AAS influences LNP-mediated mRNA delivery, the researchers assessed essential steps in LNP uptake and functional delivery – including endocytosis, endosomal escape, and mRNA translation – using a dual-labeled LNP system encoding DiD – a dye that incorporates into the particle membrane – and an EGFP-encoding mRNA. In two HPLM-conditioned cell lines, AAS increased the fluorescence of both DiD and EGFP, with the ratio remaining unchanged, suggesting that AAS likely promotes LNP endocytosis. Proteomics analysis confirmed this, showing enrichment of endocytosis-associated pathways in cells treated with AAS, while pathways associated with endosomal escape or translation were not affected. Using chemical inhibition studies with small molecules, the researchers found that AAS improved clathrin-independent carrier (CLIC)-mediated endocytosis.
The researchers then moved to in vivo assessment. Mice were injected intramuscularly, intratracheally, and intravenously with an LP01-based luciferase-encoding LNPs, with or without AAS. In vivo imaging of luciferase expression showed that at 24 hours, both groups had strong luminescence signals, but expression was significantly higher in the AAS groups across the administration routes. At 72 hours, the luminescence decreased in both groups, but the intensity remained higher in the AAS group, suggesting long-lasting effects. These effects were independent of the lipid formulation or the mRNA cargo. The effects, however, depended on timing: simultaneous administration of LNPs and AAS had the strongest effect, and delaying AAS administration reduced the beneficial effects. Additionally, effects were impacted by the AAS dose and tissue biodistribution.
Moving to therapeutic model testing, the researchers first assessed a liver inflammation model that uses acetaminophen (APAP) overdose to induce hepatocyte necrosis and inflammation, progressing to acute liver failure. Mice were injected with growth hormone (GH)-encoding LP01-LNPs, with or without AAS, 6 hours after high-dose APAP injection. GH concentrations in serum increased 8.6-fold with AAS. Further, mice receiving AAS exhibited lower serum alanine aminotransferase levels, fewer necrotic regions in liver tissue, and lower levels of proinflammatory cytokines in the liver, suggesting reduced inflammation and liver damage. Assessment of survival confirmed this, as LNPs alone improved survival and rescued 33% of mice, whereas all mice survived in the LNP + AAS group.
Gene editing is of interest for mutation-related diseases, but the efficiency of current methods varies. The researchers hypothesized that increasing intracellular expression of CRISPR-Cas9 components might improve this efficiency. To test this, hemizygous EGFP-expressing mice with widespread EGFP fluorescence throughout the body were used. A single-particle LNP was formulated with LP01-LNPs containing EGFP-targeting guide RNA and SpCas9-encoding mRNA. The LNPs were administered intratracheally, with or without AAS, and lung tissues were assessed 7 days later. LP01-LNP alone had an editing efficiency of 20-30%, while AAS enhanced it to 85-90%. Evaluation of different cell types revealed high efficiency (>90%) in endothelial and epithelial cells, whereas immune cells showed 70-80% editing efficiency, suggesting that further optimization is needed to improve efficiency in immune cells.
The data in this study demonstrate that in vivo delivery of LNPs can be improved by supplementing with amino acids. These findings, once further optimized for specific applications, could advance the clinical translation of various treatment technologies, including tumor-targeting mRNA vaccines.
Write-up by Maartje Wouters, image by Lauren Hitchings



