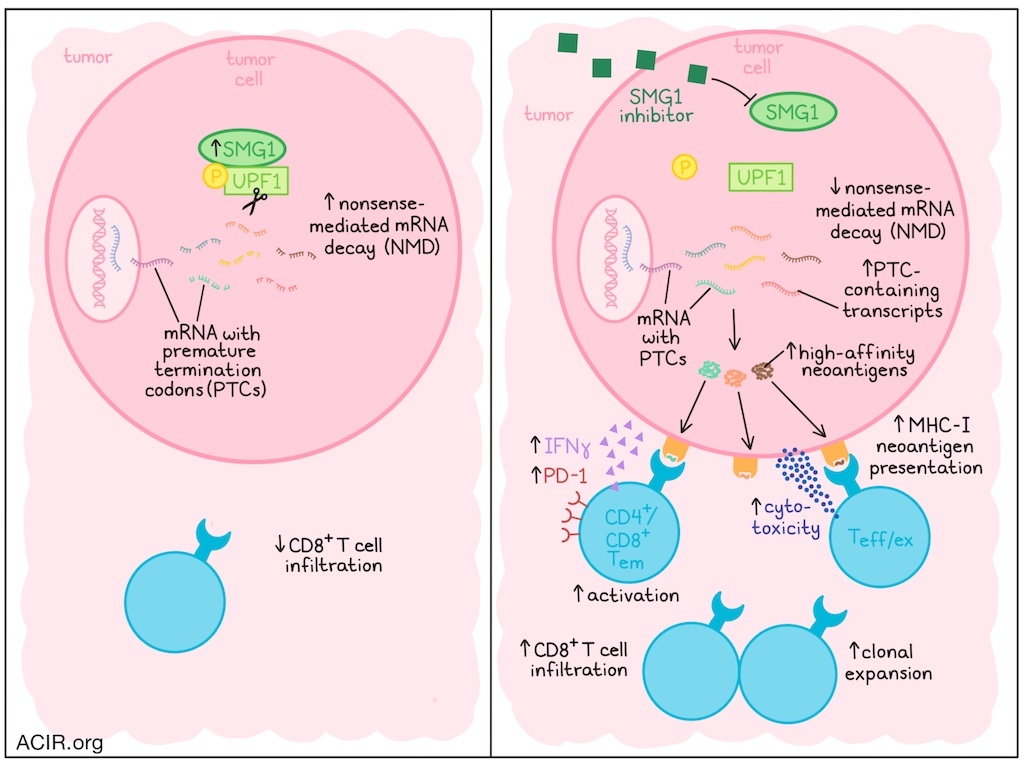
Inadequate neoantigen presentation in cancer hampers the endogenous antitumor immune response and the efficacy of immunotherapy. One mechanism that limits the production of immune-targetable antigens is nonsense-mediated mRNA decay (NMD). NMD degrades transcripts with premature termination codons (PTCs), eliminating a potential source of frameshift antigens, which may serve as an immune-evasion mechanism in tumors with elevated NMD activity. In recent work published in Immunity, Vendramin, Fu, Fernandez Patel, Zhao, et al. determined whether NMD pathway inhibition (NMDi) in cancer may promote immune-targetable neoantigen expression and presentation, and improve immune checkpoint blockade (ICB) responses.
The researchers began by assessing the contributions of NMD-related genes and the correlation between NMD components and antitumor immune responses to ICB. A comprehensive dataset of somatic and germline PTC-generating nonsense mutations was created using matched DNA and RNAseq data from 3,765 patients across 11 cancer types from TCGA and CPI1000+. An NMD score was developed, defined as the proportion of expressed PTC-containing transcripts among all PTC-inducing mutations. Lower values indicated weaker NMD activity, and higher values indicated stronger NMD activity. Lower NMD scores were associated with better clinical ICB responses, independent of tumor mutational burden (TMB) or tumor type, whereas increased NMD activity correlated with tumor progression.
A bioinformatics pipeline and scoring system were used to rank genes associated with the mechanics of NMD based on 9 parameters capturing NMD pathway relevance. SMG1, a kinase initiating the process of NMD mRNA degradation by phosphorylating UPF1, was detected as the top-scoring candidate. SMG1 protein-truncating variants (PTVs) reduced NMD activity and were associated with improved ICB responses. SMG1 expression was elevated in tumors, and negatively correlated with CD8+ T cell infiltration.
To validate the SMG1 data, the researchers generated SMG1-knockout A375 melanoma cells using CRISPR-Cas9, which resulted in complete inhibition of UPF1 phosphorylation. When SMG1 was downregulated in A375 or lung adenocarcinoma (PC-9) cells using small interfering RNAs or small molecule inhibitors, UPF1 phosphorylation was again inhibited.
The researchers determined whether small molecule inhibitors of SMG1 (SMG1i) improved immune responses in patient-derived tumor fragments (PDTFs). The cohort of PDTFs included 80% mismatch repair-proficient, microsatellite-stable (pMMR/MSS) colorectal cancers, which are characterized by low TMB and poor ICB responses. Fragments were treated with SMG1i and subjected to bulk RNAseq, which revealed upregulated PTC-containing transcripts and canonical NMD targets. In silico translation of these transcripts along with MHC-I binding prediction identified several high-affinity neoantigens, indicating that NMDi may expand the neoantigen repertoire.
In the PDTFs, NMD inhibition led to PD-1 upregulation and IFNγ secretion by effector memory CD4+ and CD8+ T cells, suggesting T cell activation. SMG1i upregulated cytotoxic genes in effector and exhausted T cell clusters, and increased NeoTCR8 (a gene signature of TCR activation) and exhaustion scores, suggestive of antigen-driven T cell activation. The exhausted compartment also showed clonal expansion, while no clonal expansion occurred among non-exhausted cytotoxic memory cells, which may be bystanders.
The researchers then asked whether SMG1i could also improve ICB efficacy. The highly aggressive, ICB-refractory LLC1 lung cancer model was treated with KVS0001 (SMG1i), anti-PD-1, or the combination. While both monotherapies had similar inhibitory effects on tumor growth, the combination worked synergistically. On day 9 post-treatment, combination therapy induced more CD4+ and CD8+ T cell activation than anti-PD-1 monotherapy, and CD8+PD-1+ T cells showed the highest level of activation. Further, SMG1i reduced overall clonotype diversity and increased the frequency of large and hyperexpanded clones.
To examine the molecular basis of SMG1i-mediated T cell activation, transcriptomic changes in A375 and PC-9 cells after SMG1i were assessed. Expression of PTC-containing transcripts was consistently upregulated, and a subset of these was detected across both cell lines. Transcript biotype analysis showed that PTC-containing transcripts mostly originated from putative NMD targets, retained introns, processed transcripts, and long non-coding RNAs. To determine whether these resulted in increased neoantigen expression, in silico translation was performed on all 8–14-mer peptides arising from the PTC-containing transcripts identified by RNAseq, and their MHC-I binding affinities were predicted. SMG1i knockdown induced a 20-fold increase in predicted MHC-I strong-binding neoantigens, up to levels detected in TMB-high tumors.
To determine if these predicted neoantigens are processed and presented by tumor cells, the transcriptomic analysis was complemented with MHC-I immunopeptidomics, in which MHC-I-bound peptides were isolated and analyzed. SMG1i increased the number of uniquely recovered peptides in both cell lines. Most of these induced peptides originated from transcripts annotated as putative NMD targets and retained introns. Eight aberrant peptides unique to SMG1i were detected, and 6 of these were predicted as strong MHC-I binders and had transcript-level induction after treatment. To determine whether these peptides were immunogenic, CD8+ T cells from HLA-matched healthy donors were primed in vitro with monocyte-derived DCs pulsed with candidate peptides. After two rounds of priming, 4/6 peptides induced IFNγ responses.
To further establish the mechanism by which SMG1i enhanced tumor immunogenicity, the researchers fused the NY-ESO-1 epitope to the NMD reporter terminus (NYE-EGFPNMD+), to allow for assessment of T cell activation upon SMG1i by comparing epitopes generated from transcripts that are NMD-targeted or that escape NMD. Stable cell lines and patient-derived lung cancer organoids (PDTO) were generated to express these reporters. Cells and PDTOs were treated with SMG1i and cocultured with CD8+ T cells expressing the 1G4 TCR, which recognizes the NY-ESO-I epitope. SMG1i enhanced T cell activation in cells expressing the reporter, and this effect was MHC-I-dependent. Live-cell imaging confirmed T cell-mediated killing of reporter-expressing cells after SMG1i.
Overall, the data from this study suggest that NMD pathway activity reduces potential neoantigen expression in tumors. Inhibition of this mechanism induces a new source of immunogenic neoantigens that can then be successfully targeted with ICB in in vivo models. If this inhibition can be safely performed in patients, this might increase the number of patients responding to ICB and other immunotherapies.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Roberto Vendramin answered our questions.

What was the most surprising finding of this study for you?
When we conceived this project, we expected that inhibiting NMD would primarily stabilize transcripts derived from frameshift indels, in line with our previous work. While this was indeed the case, we quickly realized that NMD suppression stabilizes hundreds of aberrant transcripts arising from the widespread transcriptional instability of cancer cells. These transcripts are normally rapidly degraded, yet their stabilization reveals a large, previously hidden pool of highly immunogenic peptides. The magnitude of this effect was particularly unexpected, reaching levels comparable to highly mutated tumors. This challenges the prevailing view that tumor immunogenicity is largely dictated by mutational burden, and highlights RNA surveillance as a central, targetable regulator of tumor immunogenicity.
What is the outlook?
This work opens a new conceptual and therapeutic avenue by positioning RNA quality control pathways as key modulators of tumor immunogenicity. In the near term, we aim to define the rules governing which transcripts give rise to immunogenic peptides, and to systematically map how different RNA surveillance pathways shape antigen presentation. Translationally, targeting NMD, for example via SMG1 inhibition, could sensitize immunologically “cold” tumors to checkpoint blockade, without inducing DNA damage, unlike conventional chemo- or radiotherapy. More broadly, this strategy may enable the discovery and exploitation of non-mutational antigens, thereby expanding the proportion of patients who benefit from immunotherapy.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
Focus on problems, not techniques. Early on, it is easy to define yourself by the methods you master, but impactful science comes from asking important, well-framed questions. Seek environments that challenge your thinking and surround yourself with people who are both rigorous and generous. Do not hesitate to move across disciplines if the question demands it. It is tempting to remain within your comfort zone, but following the science is more meaningful, more rewarding, and more fun! Finally, resilience matters more than speed. Progress is rarely linear, and setbacks often lead to the most interesting ideas.



