
The ACIR team attended the Keystone Symposia on Cancer Immunotherapy: Basic Mechanisms Informing Clinical Application held on March 15-18, 2026 in Quebec, QC, Canada. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below. (This Keystone Symposia conference is available for On Demand viewing! Find out more here).
Keynote address
Elizabeth M. Jaffee
Inventory of Cancer Immunotherapy
Solange Peters
Ira Mellman
Checkpoint blockade
James P. Allison
Padmanee Sharma
E. John Wherry
Cell therapies
Carl H June
Microbiome
Arielle Elkrief
Tumor immune microenvironment
Jennifer Guerriero
Hélène Salmon
Kai W. Wucherpfennig
Andy Minn
Tullia C. Bruno
Combination therapies
Sandra Demaria
Thomas Marron
Keynote Address
Priming the Immune System to Provoke Checkpoints: A Path to Maintaining the Immunotherapy Momentum - Elizabeth M. Jaffee, Johns Hopkins University
Elizabeth Jaffee opened her keynote by stressing that cancer immunotherapy, using immune checkpoint blockade (ICB), T cell engagers, and T cell-based therapies, has revolutionized cancer treatment, making 20-30% of previously fatal cancers treatable. The current focus is on expanding this success, especially for non-immunogenic cancers, like pancreatic cancer. Here, cancer vaccines can function as "extenders of efficacy" for immune modulators, including ICB, particularly those that alter the dynamic tumor immune microenvironment (TIME). Neoadjuvant clinical studies in patients with pancreatic cancer (PDAC) showed that a GM-CSF-expressing allogeneic tumor cell vaccine alone induced tertiary lymphoid structures (TLSs), but T cells remained weakly active, and PD-L1 and PD-1 were highly expressed in the TLS regions. Applying an iterative learning approach, Jaffee and colleagues showed that adding anti-PD-1 improved T cell activation, specifically increasing CD8+CD137+GZMB+ T cells, leading to a triple combination with anti-CD137 agonist (urelumab) that supercharged CD8+ T cells and induced pathologic complete responses in 3 of 10 patients. Vaccines targeting shared oncogene mutations are especially desirable, given the dependence of tumor cells on continued presence of such genes. Jaffee presented data from a study in pediatric patients with fibrolamellar hepatocellular carcinoma – a rare and aggressive cancer that is driven by the DNAJB1–PRKACA fusion gene product, an abnormal hybrid kinase protein. A synthetic long peptide (SLP) vaccine targeting the fusion gene product, combined with anti-CTLA-4 and anti-PD-1, showed a 25% durable complete response rate and 30% additional control in these patients. Reactive CD4+ TCRs have been cloned and will be developed for adoptive T cell therapy. High ammonia levels in patients with resistant disease suggested glutamine addiction of the tumor as a key mechanism of resistance to the immunotherapy, leading to a new combination trial with a glutamine inhibitor. Jaffee then presented progress in the KRAS vaccine program at John Hopkins. A 21-mer SLP vaccine targeting the six most common KRAS mutationsm, given intradermally with pICLC and with systemic anti-CTLA-4 and anti-PD-1 in the adjuvant setting to patients with PDAC, elicited mKRAS-specific responses in most patients within 17 weeks (17 weeks is time of maximal response) post vaccination. Correlative data suggested better disease-free survival in patients with a strong T cell response to their specific mutation (particularly G12V, G12R, and G12C) or to any of the mutant epitopes. Most of these cells were CD4+ central memory and CD8+ effector memory T cells. TCRseq identified mKRAS-specific CD4+ TCRs (mainly G12R), with up to 25% of these TCRs being cross-reactive towards multiple KRAS mutations. Cells with these TCRs were also cytotoxic, sometimes recognizing multiple KRAS epitopes. In patients with metastatic, chemotherapy-resistant, microsatellite-stable colon cancer, the same vaccine/ICB combination led to partial responses and evidence of vaccine-induced pre-existing and mainly de novo T cells in the peripheral blood. Interestingly, although both clinical responders and non-responders showed KRAS clonotypic expansion in the blood, only responders showed expansion of these clonotypes infiltrating the tumors. Vaccination of healthy individuals with high-risk pancreatic lesions without ICB established long-lasting immunity, with T cell responses sustained for up to two years. Samples from the majority of individuals (17 of 20) demonstrated polyfunctional CD4+ and CD8+ T cell responses against all mKRAS epitopes, and these were mainly central and effector memory T cells. Interestingly, the vaccine also induced highly activated CD8+ TEMRA cells. A new trial aims to vaccinate patients with pre-malignant cysts (PMNs) before a complicated surgery to potentially prevent progression. Jaffee concluded that while immune checkpoints have been transformative, cancer vaccines are poised to extend their value, work in non-immunogenic cancers, and are essential for tackling the global rise in pre-cancers and early-onset cancers.
Reading suggestion:
Personalized neoantigen vaccine improves anti-PD-1 in advanced hepatocellular carcinoma in early clinical trial
Inventory of Cancer Immunotherapy
Postmortem of Failed Immunotherapy Approaches - Solange Peters, Lausanne University Hospital (CHUV)
While immune checkpoint blockade (ICB) can have broad antitumor effects and has become a standard treatment for many cancers, it is only effective in a minority of patients (~20%). Solange Peters from Lausanne University Hospital reviewed lessons learned from therapy failures, comprising primary resistance, acquired resistance, and treatment-limited toxicity. She discussed the economic and healthcare context in which negative studies have emerged. Due to its efficacy across cancer types, acceptable toxicity, and long-term benefits, PD-1/PD-L1 was an attractive target for the pharmaceutical industry, resulting in strong competitive pressure for market share. However, this has resulted in the cancellation of phase 2 trials that could help detect biomarkers and confirm activity. Peters noted that approximately half of the completed phase 3 ICB trials (600-800 trials in solid tumors, $250 billion) have been negative. For anti-PD-1 and anti-PD-L1, multiple manufacturers have received market approval by conducting similar trials with comparable patient populations, which may have postponed real advances in treatment. Peters argued that several theoretical errors underlie these negative results. First is the assumption that the PD-(L)1 pathway can be successfully targeted across all cancer types, which proved false, as certain cancers (e.g., glioblastoma, prostate, and pancreatic cancer) did not respond. Second is the assumption that all pathological and clinical subgroups within a cancer type would respond equally. Biomarker-based subgrouping might improve patient selection, but it is challenging, as the more biomarkers and cut-offs considered, the more subgroups need to be studied. PD-L1 is an example of a biomarker with disease-specific utility, and where we have retrospectively learned what limits can be useful. The third error involves assuming a strategy can be uniformly applied across different clinical scenarios. For example, in ovarian cancer, frontline ICB had no clinical effect, but showed efficacy in platinum-resistant disease. As another example, the lines of treatment matter, exemplified by the frequent observation that failures or modest benefit in adjuvant therapy are frequently not replicated in the neoadjuvant setting, where significant success has been observed due to the absence of clonal divergence, immune fitness, and actionable tumor immune microenvironment (TIME). Last is the mistaken belief that novel, theoretically sound treatments, particularly synergistic combination therapies, can be directly translated into the clinic and across cancer types. For example, despite solid rationale, anti-CTLA-4 has failed in some cancers, but shown promise in others, alone or with anti-PD-1/PD-L1. TIGIT targeting is another example with a strong biological rationale, resulting in 21 companies developing inhibitors, 30 of which were assessed in 220 trials involving 50,000 patients, and costing $3.5 billion, yet none received approval. The same rush of multiple companies to participate may now be occurring with anti-PD-1/anti-TIGIT and anti-PD-1/anti-VEGF bispecifics, based on modest benefit over anti-PD-1 alone in small studies. Similarly, efforts targeting TIM3 and modalities targeting the TIME (e.g., tyrosine kinase inhibitors, IDO-targeting, cytokines) have been spectacular failures (IDO-targeting) or have shown limited efficacy, and may be effective only in specific indications (e.g., IL-15 superagonist in certain bladder cancers), or require optimization (e.g., high-dose IL-2 gave way to IL-2Rα avoidance, which is giving way to IL-2Rβ/γ targeting and other highly selectively engineered forms). Based on this, Peters questioned whether more time spent on phase 2 development could have prevented some of these negative results.
The Future of Cancer Immunotherapy - Ira Mellman, Parker Institute for Cancer Immunotherapy
Ira Mellman discussed the past and future of immunotherapy, noting that current approved modalities have been very T cell-centric, focusing mostly on modulating T cell activation and blocking exhaustion. Following the success of PD-1/PD-L1 targeting, there has been a search for additional checkpoint targets. Mellman noted that the advent of PD-1/PD-L1 immune checkpoint blockade (ICB) was an unexpected success that did not reflect long-term thinking or mechanistic understanding by the pharmaceutical industry, as it was largely an academic endeavor. Mechanistically, PD-1 is turned on by LCK phosphorylation of its cytoplasmic domain, which recruits phosphatases, including SHP2, that then activate the TCR complex or the costimulatory receptor CD28. CD28 is the preferred target, and SHP-2 dephosphorylates elements of the TCR complex only when CD28 is low. This mechanism is highly dependent on DCs, as PD-L1 expression on DCs appears to be the most critical pool for blockade efficacy. In the search for other targetable checkpoints, Mellman’s work has focused on TIGIT. TIGIT binds PVR/CD155 and activates co-inhibitory activity, but PVR can also bind CD266, with TIGIT and CD266 competing for the same ligand. Since TIGIT is the preferred binder, when it is present, there is no CD266 costimulation. This suggested that blockade of the TIGIT–PVR interaction would result in PVR binding to CD226. In murine models in which anti-PD-1 or anti-TIGIT had no effect, the combination resulted in complete remission and long-lasting antitumor immunity. Further findings revealed overlap between the TIGIT and PD-1 pathways, as the ITIM domain of PD-1 recruiting Sp2 not only dephosphorylates CD28, but also blocks phosphorylation of CD266. Therefore, there is functional overlap between PD-1, CD226, and CD28. In mice, anti-PD-L1 treatment decreased tumoral exhausted T cells and increased effector cells, while the combination almost completely blocked exhaustion. Randomized phase 2 results for this combination were promising and led to many phase 3 trials, but none have shown benefit. This lack of effect, Mellman noted, might be due to a number of factors: confounding effects of TIGIT on other cell types, the phase 2 studies having a too small sample size, mouse data not translatable, incorrect dosing, or dual inhibition might double down on a mechanism that is no longer rate-limiting in patients. This remains unclear, but investigating these questions would be important for the future of immunotherapy. Looking ahead, Mellman argued that it is important to recognize that there are many therapeutic opportunities beyond T cell exhaustion. Amplifying DC activity and addressing the tumor immune microenvironment (TIME) would be important, as these control T cell activity. Further, there is a need to focus efforts on bi- and multi-specific binders with novel mechanisms. The research requires a concerted effort to fully understand all parameters of cancer immunity. There is also a need to develop methods to assess treatment response and resistance in patients, for which AI could be helpful. Mellman then highlighted a vaccine study in collaboration with BioNTech and Memorial Sloan Kettering. Patients with pancreatic cancer treated with a personalized mRNA neoantigen vaccine after surgery could be divided into two groups: responders and rapid progressors. The progressors were more commonly splenectomized prior to vaccination, which may have contributed to the lack of response. Long-term follow-up indicated that maintenance of peripheral memory responses led to sustained antitumor activity, preventing recurrences. From various studies, it appears that vaccines exhibit greater clinical efficacy in the adjuvant setting, potentially due to lower disease burden and limited TIME-induced immunosuppression. In the metastatic setting, TIL therapy might be more successful due to polyclonal T cells being able to target tumor heterogeneity. This treatment has shown efficacy after initial ICB-induced tumor regression. Mellman finished with a summary of new technologies that may help design new immunotherapies, including new methods of mRNA synthesis, generation of “TILs” from peripheral blood, AI-driven molecular design to create antigen-specific synthetic TCRs, and in vivo engineering techniques. These new developments could extend reach to more patients, potentially lowering costs and improving scalability.
Reading suggestion:
What really happens when you block TIGIT and PD-L1 together
Checkpoint blockade
Immune Checkpoint Blockade in Cancer Immunotherapy: Mechanistic Insights Provide New Therapeutic Opportunities - James P. Allison, University of Texas MD Anderson Cancer Center
James (Jim) Allison reviewed the history and contrasting mechanisms of the checkpoint inhibitors anti- CTLA-4 (ipilimumab) and anti-PD-1 (e.g., nivolumab) to explain why these immunotherapies succeed or fail under certain conditions, and then highlighted new drug development considerations. CTLA-4 is induced as soon as a T cell receives the activation signal, and acts as a hard-wired, constitutive biological brake on the immune response, shutting off costimulation and halting T cell expansion. Early experiments in mice with anti-CTLA-4 showed dramatic tumor elimination, leading to lasting immunity. This success inspired the development of ipilimumab (ipi; anti-CTLA-4). A phase 1 trial in melanoma showed early excitement, with 3 out of 14 patients having complete responses. Later, the PD-1/PD-L1 axis was discovered, which works as an induced resistance mechanism. PD-L1 is upregulated on tumor cells in response to IFNγ produced by T cells, which then inhibits PD-1 on T cells, leading to T cell exhaustion. The 10-year follow-up data for the combined ipi and nivolumab (nivo; anti-PD-1) trial showed a remarkable survival rate of over 50% in patients with melanoma. Today, checkpoint inhibitors, primarily anti-PD-1, are frontline therapy for roughly 30% of all patients with cancer, and neoadjuvant therapy with these agents is proving even more effective. A critical comparison of the two checkpoint inhibitors reveals fundamental differences in the mechanism and clinical outcomes. While anti-CTLA-4 works during T cell priming and expands clonal diversity by lowering the activation threshold and bringing new clones into the response, anti-PD-1 works on fully differentiated T cells, expanding existing T cells that were already present, but exhausted. Anti-CTLA-4 works primarily on CD4+ T cells, but also CD8+ T cells, and can remodel the TME, particularly the myeloid-derived suppressor population with ICOS stimulation. Meanwhile, anti-PD-1 primarily affects CD8+ T cells, and does not significantly remodel the TME. After response to anti-CTLA-4, disease recurrence is rare, due to superior memory formation (TCF7+CD8+ T cells). However, for patients who respond to anti-PD-1, disease recurrence is common (25-30% of patients after 2 years), due to poor memory formation, as anti-PD-1 works only on Tox+CD8+ T cells. Both antibodies have the same general types of adverse events, but these are more frequent with anti-CTLA-4. Allison and his team showed that while anti-PD-1 monotherapy increases the number of exhausted CD8+ T cells, combination with anti-CTLA-4 leads to an elimination of these T cells and an increase in effector CD8+ and CD4+ T cells. Pam Sharma found that neoadjuvant ipi treatment of patients with bladder cancer induced the expansion of ICOShighTbet+CD4+ T cells, which produced IFNγ and so were not typical ICOS+ Tfh or Treg. These cells were critical for the antitumor response; patients whose ICOS+CD4+ counts did not stay elevated for the course of treatment did not respond. Interfering with CTLA-4 signaling revealed a more nuanced pattern of CD4+ T cell differentiation, with a host of novel phenotypes occurring. Mouse models confirmed that the ICOS/ICOS ligand pathway was required for anti-CTLA-4 to work. In a poorly immunogenic tumor model, adding an ICOSL to a GVAX vaccine dramatically increased the efficacy of anti-CTLA-4 from ~20% to 85% by increasing CD4+ TEFF, CD8+ TEM, and inflammatory myeloid cells, and decreasing TEX and suppressive myeloid cells. Allison concluded that the field must move beyond high-volume trials and focus on the underlying biology in small trials (10-12 patients). Testing molecules in combination, especially those only induced by a first-line therapy, is also essential. For instance, some molecules have been ignored because they failed as monotherapies, but may be crucial in combinations. The goal is not merely to move the median survival, but to achieve high-rate, durable responses, or cures, like those seen in melanoma with combination therapy.
Reading suggestion:
Why anti-CTLA-4 promotes better memory responses than anti-PD-1
From the Clinic to the Lab: Investigating Mechanisms of Response and Resistance to Immune Checkpoint Therapy - Padmanee Sharma, University of Texas MD Anderson Cancer Center
Padmanee (Pam) Sharma presented research focused on identifying specific mechanisms of response and resistance to immune checkpoint blockade (ICB), biomarkers that predict response, and new targets and approaches to increase the number of responding patients. Sharma is a strong proponent of neoadjuvant phase 1a/2a clinical trials to administer drugs at earlier stages of disease and potentially to treatment-naive patients, and to obtain tumor tissue to study mechanistic changes in the tumor immune microenvironment (TIME). Studies dating back to 2006, prior to the 2011 FDA approval of anti-CTLA-4 (ipilimumab), utilized this "window of opportunity" design. A 12-patient neoadjuvant anti-CTLA-4 trial in bladder cancer demonstrated early clinical signals for both safety and efficacy (3 complete responses), leading to the design of subsequent ICB trials. Dual checkpoint blockade (anti-CTLA-4 plus anti-PD-L1) in cisplatin-ineligible bladder cancer patients achieved pathological complete responses in up to 40% of patients. The analysis of the tumor samples revealed the presence of tertiary lymphoid structures (TLSs; consisting of CD4+ and CD8+ T cells, DCs, and B cells), with responders showing a significantly higher density of these lymph node-like structures within the tumor, suggesting T cell priming can occur in situ in the tumor and is not restricted to the draining lymph node. Gene expression analysis highlighted the ICOS/ICOSL pathway in these TLSs, and mouse studies confirmed that this pathway is necessary for optimal antitumor responses. Sharma and her team then explored how host genomic alterations could impact ICB response, specifically focusing on clonal hematopoiesis (CH) and how it impacts myeloid cell responses. CH, often involving somatic mutations in DNMT3A, TET2, and ASXL1 in hematopoietic stem cells, leads to a myeloid cell bias, and is common in healthy, aging individuals (over 10%) and patients with solid tumors (20%), raising the question of whether mutant myeloid cells had an impact on immune responses in solid tumors. TET2 mutations were previously linked to poor overall prognosis in solid tumors. Sharma and her team treated TET2-mutant mice with ICB, and observed significantly improved survival via an IFNγ-driven antitumor response. Mechanistically, the TET2 mutation polarized macrophages toward an antigen-presenting phenotype, driving stronger effector T cell expansion. A retrospective study of ~60,000 patients with lung or colorectal cancer confirmed that TET2 mutation carriers had better overall survival when treated with ICB, but not with traditional frontline therapies (chemotherapy, TKI). This work established TET2 CH as a biomarker for selecting patients who will benefit from anti-CTLA-4 therapy, as this was seen to be predominantly an effect of anti-CTLA-4, and not anti-PD-1. Sharma closed by again emphasizing the importance of small neoadjuvant trials to develop direct mechanistic information critical to later drug development.
Reading suggestion:
Macrophage makeover required for effective CTLA-4 and ICOS combination therapy
Digesting TEX Progenitor Biology Reveals New Targets for Combination Immunotherapy - E. John Wherry, University of Pennsylvania
John Wherry focused on the question “How do we drug exhausted T cells?”, as these cells are “the point of the spear” for most immunotherapies. Compared to memory T cells, characterized by high functionality, effective recall, and independence from the key transcription factor TOX, exhausted T cells (TEX) have poor, but still some functionality. They require ongoing TCR stimulation for maintenance, and are dependent on TOX. Wherry argued that the several subsets of TEX cells along the differentiation axis, all of which have a similar “exhausted epigenetic state” are very different from memory populations and function like a tissue, with self-renewing progenitor exhausted-like stem cells differentiating through an active proliferation phase, and finally taking up residence in inflamed tissues and tumors in their terminal state. The progenitor cells (Tpex) are the target of PD-1 blockade, producing the critical proliferating intermediate effector-like cells. Ly108 (SLAMF6) and CX3CR1 serve as markers that distinguish these populations. Genetic knockout of PD-1 is known to affect accumulation of TEX, and so Wherry posed the question as to why we are clinically blocking PD-1, essentially permanently, instead taking the perspective that we shouldn’t be focusing on drugging PD-1, which is just the pro-drug, but instead on the pharmacology of the drug, which actually is the T cell. Rapidly progressing animal models are challenging to do extended pharmacology experiments, don’t allow the study of the pseudo-equilibrium state characteristic of most human tumors, and are plagued by antibodies quickly developing against rat anti-PD-1 antibodies. After converting a rat to a mouse anti-PD-1 antibody, Wherry was able to investigate alternative dosing regimens. In the chronic LCMV model, mimicking long-term pseudo-equilibrium, comparing a single 2-week cycle of anti-PD-1 to continuous 3 months anti-PD-1 treatment showed there was no added benefit of continuous treatment in the longer-term number of circulating viral-specific T cells. This was similar to studies in humans showing only a single early burst of TEX reinvigoration following anti-PD-1, despite long-term treatment. In a slower-growing tumor model (MCA1956), continuous blockade was no more beneficial than a one week cycle. Wherry then introduced the concept of a “drug holiday” – an interruption of continuous dosing to allow the system to reset and again be responsive to treatment. In the LCMV model, this led to better long-term T cell numbers and better disease control across multiple tissues. Qualitatively, continuous treatment reduced the proportion of the intermediate effector-like cells, while the drug holiday maintained this key population. Investigating why, Wherry showed that long-term treatment actually limited the capacity of cells to respond proliferatively to anti-PD-1, and drove differentiation toward the terminal, ineffective state. Transcriptional profiling showed these long-term treated cells were driven to a single state characterized by induction of stress responses and an inability to proliferate. Interestingly, CD22, a classic B cell inhibitory molecule, which controls BCR signaling as a survival mechanism, was uniquely expressed in a subset of Tpex cells after long-term anti-PD-1 treatment. Adoptive transfer of isolated CD22+ Tpex into late chronically LCMV-infected mice (to synchronize the infection) were more proliferative than CD22- Tpex. However, similar transfer of CD22+ Tpex cells following long-term anti-PD-1 treatment led to CD22+ Tpex cells with more limited differentiation toward the intermediate effector-like cells. Adding CD22 blockade, as a separate antibody or as part of a bispecific anti-CD22/anti-PD-1 construct, either at the end of long-term anti-PD-1 treatment or with up-front, short-term anti-PD-1, led to recovery of the intermediate effector-like cells and better viral control. Experiments to date have indicated that B cells are not involved in these effects. In the AT3-OVA tumor model, the bispecific drug improved tumor control compared to either antibody alone. Wherry closed by summarizing a variety of data indicating that timing of stimulation of T cells, and importantly rest between stimulations, have very important effects on the quality and efficacy of progenitor cells.
Reading suggestion:
Checkpoint receptors - unexpectedly essential for forming memory T cells
Cell therapies
Strategies to Adapt CAR-T Cells for Solid Tumors - Carl H June, University of Pennsylvania
With a backdrop of watching the over 40-year path from the discovery of the Philadelphia chromosome to the approval of the first BCR-ABL targeting precision therapy imatinib, and being one of the leaders in the development of CD19 CAR therapy from discovery to FDA approval in just 20 years, Carl June provided an overview of CAR T cell therapy challenges for solid tumors, which is hoped, despite the challenges, will proceed even faster due to technological advancements and generated knowledge. One example is learning about the cell-extrinsic factors that can impact design. Analysis of T cells from patients with sustained B cell aplasia and leukemia remission 8 years after therapy revealed the criticality of type 2 cytokine-secreting CD4+ T cells to prevent late exhaustion of CD8+ T cells, emphasizing the unappreciated importance of cross-talk between cells in the CAR T cell product. A major challenge for CAR T cells is maintaining activity in the face of multiple suppressive challenges in the solid tumor immune microenvironment (TIME). Armouring CAR T cells with a variety of orthogonal cytokines can help to maintain the persistence and function of CAR T cells. One example is adding IL-15, which can improve persistence, and has generated exciting data in pediatric solid tumors, with ongoing studies adding both IL-15 and IL-21. June described his team’s work with IL-18 armoring, giving these cells the ability to both reprogram the TIME and to enhance CAR function, as IL-18R is expressed in T cells. Lymphoma patients who had failed prior cell therapy showed an 80% overall response rate with CD19_IL-18 CAR-T. Studies are ongoing in solid tumors. These examples highlight the importance of both thinking about cell-intrinsic approaches to improve CAR-T (armoring, genome editing, transcription factor disruption) and cell-extrinsic approaches (targeting cell or molecular barriers in the TIME). IL-9 is a particularly interesting armoring agent, as although T cells express IL-9, they do not express the IL-9 receptor. By armoring CARs with IL-9R, this creates an orthogonal receptor–cytokine pair that is highly specific only for the engineered CAR T cells. In mouse models, a mesothelin (MSLN)_IL-9R CAR + IL-9 was 30-fold more potent, and also showed enhanced activity in a prostate cancer model. Armoring with IL-9R appears to work through broad STAT1, 3, 4, and 5 signaling, and through reduction of ATF4 (a master transcription factor for the stress response) delaying exhaustion in CD8+ T cells. June then turned to vignettes for some additional encouraging results for CAR-T in solid tumors. The CAR most advanced in clinical development targets Claudin-18 isoform 2, which has completed a phase 2 randomized study in advanced gastric and gastroesophageal junction cancer. Despite achieving statistical significance versus physician’s choice of standard-of-care, the improvement in overall survival was only 2 months, so still a way to go. June has worked on MSLN-specific CARs for over 15 years. Mesothelin has also been approached with antibody–drug conjugates and with bispecific T cell engagers. Early targeting agents initially recognized the N-terminus of the extracellular domain, but proteolytic cleavage of this region led to shed target, which abrogated activity. Later, scFvs were developed to target the remaining “stump”, and these are now in clinical development. MSLN targeting was initially stalled due to binder-dependent toxicity, and a murine toxicity model was developed that allowed identification of a dose with less on-target, off-tumor toxicity. One patient with pancreatic cancer (PDAC) in a MSLN-CAR trial was an example of another challenge – CAR T cell penetration into the tumor. For this patient, multiple sites of metastatic disease rapidly cleared, but the primary tumor progressed. Histologically, a lung metastases from another patient showed a defined ring of T cells surrounding, but not penetrating the tumor. One approach to enhance penetration is to eliminate the cells that create physical barriers, like cancer-associated fibroblasts, which can be targeted with an anti-fibroblast activation protein (FAP) CAR. In animal models, treatment with anti-FAP CAR-T followed by a MSLN-specific CAR-T significantly extended survival in a PDAC model. June then described efforts to overcome induction of exhaustion by conducting transcriptomic and epigenetic analysis of in vitro repeated stimulation models and clinical CAR T cells isolated from treated patient liver biopsies to molecularly dissect the root causes. The transcription factors SOX4 and ID3 emerged in both sample sets. Finally, June wrapped up with an early look at dual-targeting CARs (EGFR and IL-13Rα2) in GBM, with some exciting, but early clinical readouts.
Reading suggestion:
IL-9-powered CAR-T for improved solid tumor responses
Microbiome
The Gut Microbiome as a Therapeutic Target in Immuno-oncology: From Fecal Microbiota Transplantation to Next-Generation Microbiome Agents - Arielle Elkrief, Université de Montréal
The gut microbiome is a critical mediator of the cancer-immune set point, with trillions of bacteria and 100 times more genes than the human body. Landmark studies showed that an intact gut microbiome is required for immunotherapy response, establishing it as a hallmark of cancer. Arielle Elkrief and her team used shotgun metagenomics to identify beneficial bacteria (e.g., Faecalibacterium prausnitzii, Akkermansia muciniphila) and deleterious bacteria (e.g., Enterocloster) associated with immunotherapy response. A major translational finding is that antibiotic use, before or during immunotherapy, significantly worsens overall survival. Antibiotics cause a bloom of bad bacteria, like Enterocloster, leading to the loss of MAdCAM1 in the small intestine, which allows immunosuppressive Tregs to enter the tumor, causing resistance. Studies in patients with refractory melanoma using fecal microbiota transplantation (FMT) from patients who had a partial or complete response to anti-PD-1 showed encouraging response rates (20-30%). In the frontline setting with healthy donor FMT and anti-PD-1, response rates were 65%, higher than the 40% ORR expected with anti-PD-1 alone. The phase 2 healthy donor FMT and anti-PD-1 LUMINate trial showed an 80% response rate in patients with NSCLC and, with additional anti-CTLA-4, 75% in patients with melanoma. Analysis of the FMT trials revealed that clinical response was not predicted by strain sharing or overall similarity to the donor. Instead, responders showed a greater elimination of their baseline, pre-existing bacteria – a finding confirmed by multiple techniques. The eliminated bacteria included harmful, immunosuppressive organisms like Enterocloster. Importantly, the overall count of different species in the sample (alpha diversity) remained stable in the responder samples, because donor or new species occupied the resulting "empty spots". In vivo evidence confirmed that re-introducing these lost bad bacteria reverses the antitumor effect of checkpoint blockade and FMT in mice. Metabolomic analysis showed that responders had decreased levels of tryptophan downstream metabolites (kynurenic acid, kynurenine), which inversely correlated with bacterial elimination. Immunologically, elimination correlated positively with activated CD69+CD8+ T cells and negatively with Tregs. An independent randomized FMT trial in renal cell carcinoma also confirmed that greater baseline bacterial elimination correlated with response, but only in the FMT group. Elkreif and colleagues have now launched a randomized phase 2 trial of FMT and checkpoint blockade in patients with metastatic melanoma. However, FMT has scalability and safety limitations. In the anti-CTLA-4 cohort of the LUMINate trial, a higher-than-expected rate of myocarditis was observed, and the onset of Grade 3 or higher diarrhea/colitis was accelerated. This toxicity was linked to donors, and subsequently the recipient patients, enriched with Prevotella bacteria. To address FMT limitations, research is focusing on scalable strategies, such as the prebiotic Camu Camu extract from a Brazilian berry. The active ingredient, castalagin, showed activity in reversing anti-PD-1 resistance in a breast cancer model. A phase 1 trial of Camu Camu plus checkpoint blockade in patients with refractory melanoma showed an encouraging safety profile, a 20% overall response rate, and a 1-year overall survival of 57%. Elkrief concluded that both FMT and the Camu Camu prebiotic strategy offer strong proof of concept for modulating the gut microbiome in cancer therapy. The future, according to Elkrief, involves developing targeted live biotherapeutic products and customizing microbiome therapies based on the patient's level of dysbiosis.
Reading suggestion:
Good things come in threes: results from clinical trials to improve checkpoint blockade
SITC - 40th Anniversary Annual Meeting 2025 (Presentation by Bertrand Routy)
Tumor immune microenvironment
From Complexity to Therapy: Targeting Tumor-Promoting Lipid Macrophages - Jennifer Guerriero, Brigham and Women's Hospital
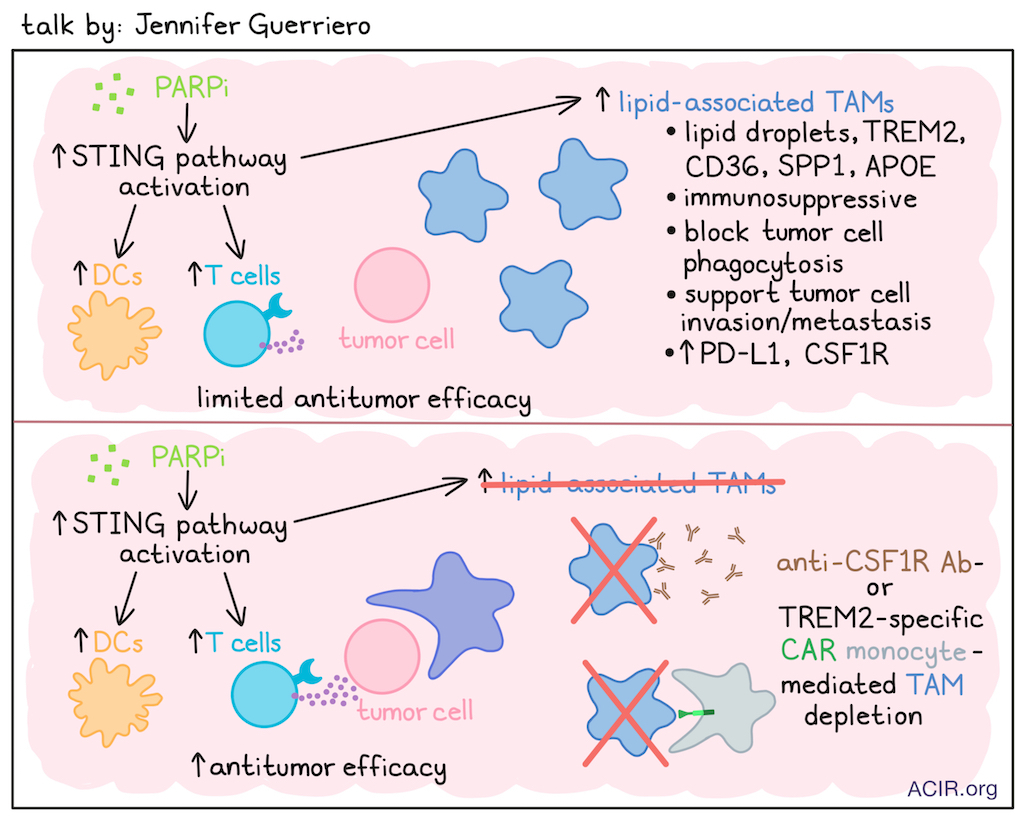
Jennifer Guerriero’s work has focused on characterizing tumor-associated macrophages (TAMs) across breast tumors, where elevated TAM levels are associated with worse outcomes across multiple tumor types. Breast tumors recruit large numbers of monocytes, and support their differentiation into macrophages, which acquire a phenotype that supports cancer cell survival, angiogenesis, and metastasis, and is highly immunosuppressive. Therefore, targeting TAMs is of interest, but, so far, has not yielded major successes. A better understanding of TAM complexity (phenotype, spatial organization, metabolic programming, homeostatic tissue regulation) might reveal more effective therapeutic targets. Therefore, Guerriero and team generated a scRNAseq TAM atlas to characterize the different phenotypes in the breast TME, revealing 10 subtypes. (Guerriero digressed briefly to urge the field to move beyond the uninformative M1/M2 description toward a more exact description of particular subtypes.) GPNMB TAMs (TREM2, APOE, FABP4) and SPP1 TAMs (CD73, MARCO, TREM2) were most frequently found across breast tumors. GPNMB TAMs exhibited a metabolic lipid signature, and SPP1 TAMs exhibited lipid signaling and hypoxia signatures. Lipid-associated TAMs contain lipid droplets and express TREM2, CD36 (lipid uptake receptor), SPP1, and APOE; they are found in normal tissues but are more abundant in tumors, where they are highly immunosuppressive, block tumor cell phagocytosis, and enhance invasion and metastasis. In a murine TNBC model, PARP inhibitor (PARPi) treatment induced suppressive lipogenic PD-L1+CSF-1R+ TAMs. In vitro, PARPi increased macrophage CSF-1R and PD-L1 expression, with transcriptomic differences in lipid metabolism. In work that had followed a separate path demonstrating that PARPi treatment led to upregulation of the STING pathway and strong T cell immunity, multiple clinical trials combining a PARPi and anti-PD-L1 were conducted, but none showed clinical benefit. Guerriero then asked whether this was due to PARPi also inducing immunosuppressive TAMs. This was investigated using samples from patients with advanced BRCA-mutated breast cancer treated with PARPi plus ICB in a clinical trial. PARPi induced STING signaling, increased T cells, DCs, and suppressive TAMs with lipid metabolic signatures. To target these TAMs, anti-CSF-1R therapy was assessed in a murine model. Combining this therapy with PARPi improved survival, which was dependent on CD8+ T cells, as was predicted based on the activities of the individual agents. This led to a phase Ib trial of the approved anti-CSF-R1 drug axatilimab in combination with olaparib (PARPi) in patients with metastatic HER2-negative BRCA-associated breast cancer; this trial is currently enrolling. Looking for other targets, TREM2 was commonly detected on these suppressive, lipogenic TAMs. In preclinical models, TREM2 deletion or inhibition is antitumorigenic, and dual anti-FOLR2 and anti-TREM2 CAR-T can clear TREM2+ TAMs and induce tumor regression. Given the unsatisfactory performance of CAR-T cells targeting solid tumors, for a variety of reasons, such as poor infiltration into the tumor, as an alternative, monocytes were engineered to express an anti-TREM2 CAR. Upon differentiation into macrophages in the tumor, these TREM2-CAR_Mono can phagocytose TREM2+ TAMs. Phagocytosis of target cells was confirmed in vitro. In the in vivo E0771 model, which has many TREM2+ macrophages, treatment with these CAR-monocytes doubled CD8+ T cells after 2 weeks, and reduced tumor burden by 50%. The effects could be further enhanced by combining treatment with anti-PD-1. Current work focuses on bringing this therapy to the clinic and researching improvements to the intracellular domains and cytokine armoring.
Reading suggestion:
Focus on suppressive macrophages may boost effects of PARP inhibitors
Cancer-Associated Fibroblasts (CAFs) as Regulators of T Cell Exclusion and Tumor Invasiveness - Hélène Salmon, Institut Curie
Hélène Salmon discussed cancer-associated fibroblasts (CAFs), which, compared to regular fibroblasts, are more activated, express more matrix proteins, and exhibit altered cytokine and other soluble factor gene expression profiles. By immunohistochemistry, CAFs can form capsules of collagen surrounding tumor nests, preventing T cell infiltration. In bladder cancer, there is a need to predict and prevent disease progression of non-muscle-invasive bladder cancer (NMIBC), the stage where most patients are diagnosed, to muscle-invasive bladder cancer (MIBC), which has a challenging prognosis. Comparing various cell types in the tumor immune microenvironment (TIME) between NMIBC and MIBC revealed that fibroblasts were most distinct between the stages. One fibroblast subtype was mostly observed in NMIBC and expressed ITGA8 and F3, and these cells were transcriptionally similar to those found in healthy bladder tissue. At later stages of disease, activated FAP-expressing fibroblasts were observed, and their proportion increased with cancer progression. The presence of these FAP+ CAFs in T1 NMIBC tumors was associated with progression to MIBC. Spatially, these CAFs were mostly detected surrounding small tumor clusters (the presence of such clusters pathologically define the T1 stage, which has a 15-20% rate of progression to MIBC). Tumor cells in the clusters surrounded by FAP+ CAFs expressed F3 (tissue factor, CD142), and had an invasive and epithelial-mesenchymal transition phenotype. Similar F3-expressing tumor cells surrounded by FAP+ CAFs have also been observed in NSCLC. In various in vitro assays using human NMIBC tumor cell lines, the presence of FAP+ CAFs enhanced tumor cell invasiveness and migration, which was dependent on F3 expression. FAP+ CAFs were found to secrete HGF and several proteases, and migration effects were partially induced by MMP, HGF, and MET. In a cohort of patients with NMIBC T1, the presence of both F3+ tumor cells and FAP+ CAFs predicted worse prognosis, identifying this subgroup as high-risk for progression to MIBC. Salmon noted that targeting CAFs is challenging, but several strategies have been proposed, including CAF depletion, CAF repolarization, extracellular matrix targeting, and targeting of downstream effectors. While preclinical work has shown the success of such strategies, clinical trials, primarily in PDAC, have so far been disappointing, but also lack in studies evaluating the mechanism of action. Given the known role of TGFβ in inducing CAFs, Salmon and team participated in a window-of-opportunity (W-O-O) neoadjuvant phase 2 trial to assess 6 patients with head and neck cancer who received 2 injections of bintrafusp-α (BA), a bifunctional fusion protein targeting both TGFβ (trap) and PD-L1 (ICB), with 4 patients having a pathological response. Treatment upregulated IFNγ-related genes and strongly reduced TGFβ signaling, particularly in stromal cells. Further, there was rapid remodeling of the fibrillar architecture, a decrease in particular collagen forms, and a reduction in activated LRRC15+ myCAFs, while iCAFs were not impacted. Similar results were also observed in another small W-O-O trial. Transcriptomics data suggested CAF depletion rather than repolarization. Histologically, the pre-treatment indication of CD8+ T cell exclusion in tumor nests was reversed, and a major responder in this cohort (54% pathological response) had higher tumor mutational burden and PD-L1 expression, and was the only patient with lymphoid aggregates in pre-treatment samples. Salmon concluded that the possible downsides of CAF targeting need to be investigated, as it can cause major stroma loss, impacting blood vessels (bleeding is a common AE of this drug) and potentially tumor dissemination.
Reading suggestion:
REVIEW: Host tissue determinants of tumour immunity
Spatial Analysis of T cell-mediated Immunity in Human Cancer - Kai W. Wucherpfennig, Dana-Farber Cancer Institute
Kai Wucherpfennig presented data on the spatial analysis of immune and tumor cells in patients with recurrent glioblastoma (GBM) who received a single injection of oncolytic virus (oHSV) as part of a clinical trial. Using CODEX with GBM tumor, immune, and stromal panels, and Xenium with a custom immune-oncology panel of gene targets (e.g., GBM, TCR, oncolytic virus), the researchers performed spatial analysis of immune–tumor interactions on tissue obtained before and after treatment. Imaging showed a dense infiltration of T cells following a single oHSV treatment. Distinct areas were observed in the TME, including zones with extensive contact between tumor and T cells, and areas with poor T cell infiltration. At both early and late timepoints, very high infiltration was detected, suggesting a persistent effect. Granzyme B (GzmB)-expressing CD8+ T cells were found in direct contact with tumor cells expressing cleaved caspase-3, indicating tumor cell killing. The density of cleaved caspase-3-positive tumor cells positively correlated only with GzmB-positive T cells, and the patterns of gene expression in tumor cells close versus distant to T cells were distinct, and indicative of immune reactivity (MHC-I, PD-L1). Patients with tumors containing GzmB+ T cells close to dying tumor cells had longer progression-free survival. Xenium analysis revealed that CD8+ T cells in highly infiltrated areas and in close proximity to tumor cells expressed a tissue-residency program (including ITGAE [CD103] and ZNF683 [Hobit]), which may help T cells persist, and were in an early activation state, expressing genes under direct TCR control (NR4A1, CD69, IFNG, TNF). Tumor-proximal T cells also expressed chemokines that recruit T cells to the tumor. There were also cells expressing memory markers (TCF7, IL-7R), which resided mainly in lymphoid clusters. These clusters contained stem-like CD8+ T cells, B cells, and DCs. Overall, T cells with an effector/cytotoxicity program were detected deep in the tumor, and in other areas, these lymphoid cluster reservoirs were detected with T cells expressing chemokines (CXCL13, CCL19, CCL21) that may help maintain this immune response. At later timepoints, the oncolytic virus was restricted to necrotic tumor regions, where macrophages and neutrophils, but not T cells, also resided. Therefore, the residual virus may not be driving the ongoing T cell response. Studying this further with TCRseq revealed sets of clones expanding in the tumor that were present prior to treatment, and these clones were often found in close contact with tumor cells. The expansion of these pre-existing clones is observed in some patients, whereas in others, they disappeared after treatment. Patients with clonal expansion had longer survival. Mapping these TCRs against known sequences showed that the vast majority were not found in public databases, and the very few that were were not HSV-reactive. An association was found between clonal contraction and dexamethasone treatment, suggesting that this treatment should be considered in future immunotherapy trials. Assessing mechanisms of resistance revealed tumor cells in a hypoxic state at a distance from T cells, suggestive of hypoxic cores in the tumor resistant to T cell infiltration. Finally, Wucherpfennig discussed how spatial analysis can be used to investigate immune pathways in cancer, using the CD161/CLEC2D pathway as an example. Spatial analysis showed high levels of CLEC2D expression in tumors, with the highest levels in hypoxic cancer cells and immunosuppressive myeloid cells.
Reading suggestion:
Resident memory T cells are ready to respond in neoadjuvant checkpoint blockade
Chronic Interferon, Inflammatory Memory, and Immunotherapy Resistance - Andy Minn, Memorial Sloan Kettering Cancer Center
Andy Minn discussed the role of inflammation in cancer growth, suggesting that understanding the immune system’s evolutionary mechanisms for dealing with threats without harming the host can contribute to development of better strategies for cancer treatment. Current “more is better” inflammatory cancer immunotherapies, such as immune checkpoint blockade (ICB), cell therapies, and cytokines, can lead to active, but often not durable responses (as with anti-PD-1 therapy), due to compensatory mechanisms for chronic conditions to protect the host, generating a host-pathogen stalemate. On the immune cell side, this includes focusing T cell differentiation toward the exhausted state to support chronicity. While less is specifically known about adaptations on the host side to respond to a chronic problem, these fit under the general rubric of inflammatory memory. An example is the adaptation of intestinal epithelium to inflammatory bowel diseases by enhancing differentiation toward secretory versus absorptive cells. As interferon (IFN) plays opposing roles in both immune and host inflammatory memory, a key question for Minn is where and when along the spectrum of inflammation should we land. In fact, prior work in chronic LCMV showed persistent stimulation of an undesirable subset of IFN-stimulated genes (“bad”-ISGs) and that blocking IFN signaling improved T cell functionality, lymph node structure, and control of longer-term infection. To adapt this to cancer, Minn focused on evaluating tumor cells that were surviving after first-round ICB in mice, and showed that these cells also upregulated these bad-ISGs, leading eventually to exhausted T cells and a myeloid-rich tumor immune microenvironment (TIME). Analysis of human samples also showed a correlation between bad-ISGs, T cell exhaustion, and outcomes, rising to the level of a commonly found metaprogram contributing to intratumoral heterogeneity. Spatial analysis demonstrated that colocalization of tumor cells expressing these ISGs and exhausted T cells or suppressive macrophages correlated with poor response to chemotherapy plus ICB. To account for all this data, Minn proposed a cellular and molecular feed-forward mechanism, beginning with a subset of cancer cells that respond to chronic IFNγ by undergoing epigenetic reprogramming, initiating an inflammatory memory state sustained by epigenetically induced upregulation of endogenous retrovirus elements, and leading to chronic virus mimicry and chronic type I IFN (IFN-I). IFN-I leads to upregulation of bad-ISGs, which restrict tumor control in multiple ways and also enhance the exhausted state of T cells, reinforcing this loop through chronic IFNγ. IFNγ exposure of tumor cell lines resulted in cells now resistant to ICB in mice, just like cells derived from late relapsers. The chromatin states of these cells were altered, and expression of bad (aka “memory”)-ISGs was sustained following IFNγ washout. Knockout of the RNA sensor MDA5 (or combined inhibition of JAK1 and TBK1) supported the role of endogenous retrovirus elements as “chronic virus” in this process. Analysis of chromatin states in cells resistant to ICB showed that JAK1 or JAK1 plus TBK1 inhibition led to cells less likely to adopt the resistant chromatin states, an erasure of immune memory. Within these ‘immune memory-erased’ tumors, TAMs were decreased, stimulatory markers on DCs were increased, and CD8+ T cells moved away from a terminally exhausted profile and toward a TCF-1+ progenitor (Tpex) profile, culminating in better tumor control. In a trial of patients with NSCLC in which an “anti-inflammatory” JAK1 inhibitor interrupted “inflammatory” anti-PD-1 treatment, appropriate changes in the proliferation of exhausted and non-exhausted T cell populations were observed in some patients in correlation with JAK1 inhibitor therapy. Importantly, clinical responses were seen even in patients who did not show a proliferative response in exhausted cells following the initial anti-PD-1 therapy (and who would thus not have been expected to respond, based on the data with anti-PD-1 alone). Minn closed by suggesting the importance of balancing the strong inflammatory therapies, like anti-PD-1, with temporary anti-inflammatory therapies, such as JAK1 inhibitors, to allow the immune system a period of “rest and recovery” and perhaps erasure of inflammatory memory in immune and non-immune cells, so that future therapy can be more effective.
Reading suggestion:
JAK-ing up ICB responses with JAK inhibitors
Unleashing the Anti-Tumor Potential of B Cells and Tertiary Lymphoid Structures for Improved Immunotherapeutic Strategies - Tullia C. Bruno, University of Pittsburgh
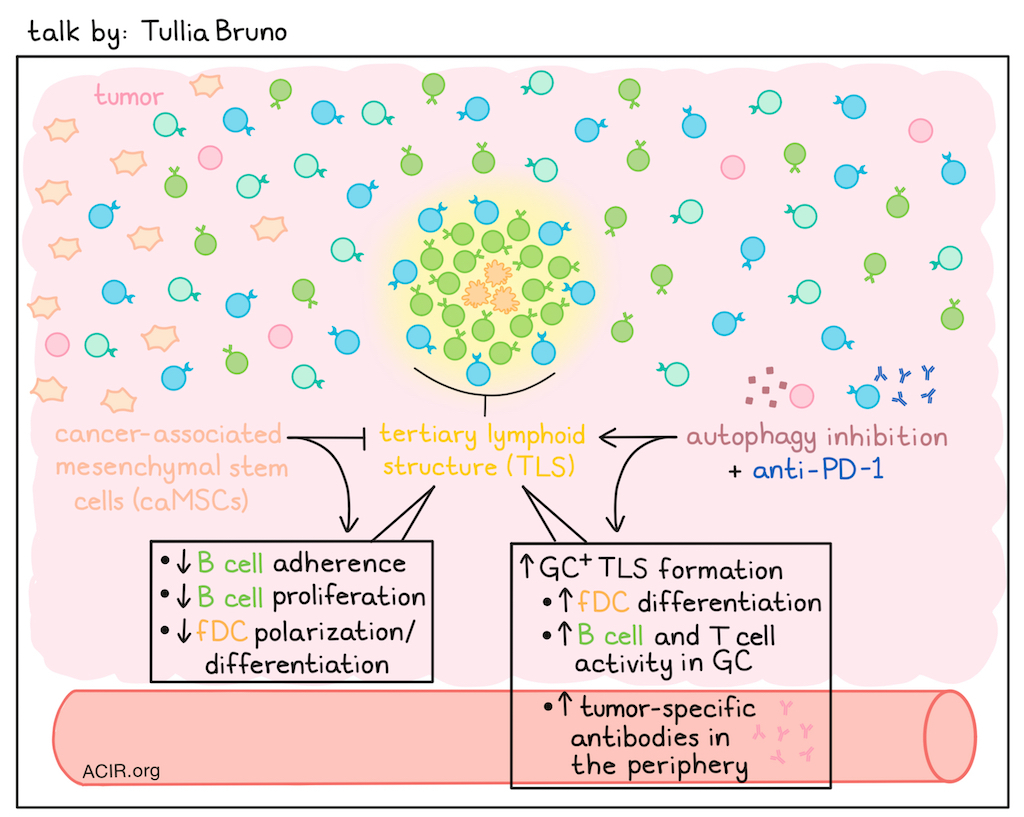
Tullia Bruno’s research focuses on tertiary lymphoid structures (TLSs), their formation and function, and whether they can be targeted. TLSs require multiple cues to form (chemokines, lymphotoxin B, antigen, the right type of stroma, etc.) and are not a uniform state in solid tumors. Rather, they exist along a spectrum with three main states, ranging from lymphoid aggregate, to TLS, to TLS with a germinal center (GC) – a spectrum that has been refined using advanced spatial biology. GC+ TLSs are less common, vary across cancer types, and are rarely present in the treatment-naive setting. As an example, comparing HPV+ and HPV- head and neck squamous cell carcinoma (HNSCC) revealed differences in B cell populations and TLS and GC formation; however, TLS activity, as measured by Ki67 and antibody maturation, and its prognostic value were similar across these cancer types. In patients with ovarian cancer, however, differences were found between tumors in the fallopian tube, omentum, or ovary, with tumoral TLSs in the ovary having reduced GC formation and B cell activity. In brain metastases of lung cancer or melanoma, TLS activity could be very different. Bone marrow-derived or normal mesenchymal stem cells (MSCs) migrate to inflamed areas, and are precursors of follicular DCs (fDCs), while cancer-associated MSCs (caMSCs) are “educated” by the tumor, promote tumor initiation, and are immunosuppressive. Bruno and team developed a caMSC signature whose expression increased with increasing distance from TLSs. In vitro testing of caMSC revealed lower B cell adherence, reduced B cell proliferation, and inhibited fDC polarization. Patients with ovarian cancer expressing both caMSC and TLS signatures had survival outcomes similar to those without the TLS signature, whereas those with only the TLS signature had better outcomes. In pancreatic cancer, fDC differentiation is negatively impacted by the presence of caMSCs. In clinical trial samples, autophagy inhibition plus anti-PD-L1 increased fDC differentiation in TLS, while both monotherapies and the combination increased B and T cell activity within the GC, resulting in higher peripheral levels of tumor-specific antibodies. Therefore, GC could be induced by therapy, depending on the therapeutic and the microenvironment. Intratumoral B cells that are not in a TLS and are in contact with tumor cells become exhausted, with reduced effector function, decreased plasma cell differentiation, and increased expression of inhibitory and homing receptors. The inhibitory receptor FcRL5 inhibited intratumoral B cell activity, and in vitro FcRL5 blockade increased B cell proliferation in the absence of tumor cells. In the presence of HNSCC cells, treatment was only beneficial when those tumors were HPV+ and TLR9 was present. Bruno and team aimed to further develop murine models to study TLS. One model is an orthotopic carcinogen-induced model, which results in non-small cell lung cancer (NSCLC)-like lesions. In this model, TLSs take 10-15 weeks to form, and may decrease in later stages of disease. These TLSs may contain GCs, and tumors with GC+ TLSs have lower tumor burden than those without GCs; TLSs without GCs were prognostically beneficial only when they were active TLSs. Tumors without TLSs had a lower tumor burden, but were characterized by strong oncogenic pathways, suggesting growth potential, whereas tumors with GC+ TLSs exhibited upregulated inflammatory pathways. B cell depletion reduced TLS formation, and those that formed lack GCs, resulting in reduced peripheral antibody levels and a higher tumor burden. Searching for therapeutic targets and biomarkers uncovered CD52 and CD37, which are B cell-specific genes associated with more active TLSs. Low expression of these genes was associated with worse survival in NSCLC. Finally, a phase 2 clinical trial in patients with metastatic melanoma showed that anti-LAG3 helped B cells remain activated, and anti-LAG 3 plus anti-PD-1 increased plasma cells. These data suggest these structures can be therapeutically targeted.
Combination therapies
Priming the Immune System: In situ Vaccination - Sandra Demaria, Weill Cornell Medical College
Sandra Demaria, building on a theme of the importance of T cell priming, focused on in situ vaccination strategies, which use the antigens present in the tumor itself for priming. Radiation – which can induce immunogenic cell death, attracting and activating DCs for antigen uptake, migration to draining lymph nodes, and presentation to T cells – can be a key initiating factor, but requires other therapies, such as immune checkpoint blockade, to be fully effective. Clinical studies combining radiation with either anti-CTLA-4 or anti-PD-1 have shown immunological effects, including induction of neoantigen-specific T cells, de novo response, and clonotypic expansion, but clinical effects have been limited, leading to a search for barriers and approaches to overcome them. Radiation works by activating a canonical pathway of anti-viral defense through release of mitochondrial DNA into the cell cytoplasm, and activation of the cGAS-STING pathway, resulting in the production of type I interferon (IFN-I; a potent activator of DCs). This innate immune pathway, though, is often downregulated by tumors, particularly at the transcription level by epigenetic downregulation of STING. However, cGAMP can be exported from the cell and activate the myeloid compartment, including DCs. Tumors can block this pathway through the action of the extracellular nucleotidases converting cGAMP to AMP (via ENPP1) and then to adenosine (via CD73), eliminating the stimulatory cGAMP and creating inhibitory adenosine. In the TNBC 4T1 model, these cGAMP-degrading pathways are active, but a combination of radiation, CTLA-4 blockade, and CD73 blockade led to significant tumor control and a reduction of non-irradiated lung metastases. After observing that adenosine signaling is very high in rectal cancer, and based on successful studies of CD73 blockade in rectal cancer models, a clinical trial employing a dual-antagonist of the A2aR and A2bR adenosine receptors, short-course radiation, and then consolidation chemotherapy, anti-PDp1 and the dual-antagonist was initiated in treatment-naive patients. Clinical results were very encouraging, with a higher rate of pathological complete responses than historical controls. Demaria then focused on how to modulate DCs, the key player in initiating T cell responses. A key inhibitory pathway in uptake of tumor cells and cell debris is the CD47 (“don’t eat me”)/SIRPα pathway, and blocking this pathway can improve tumor control in some models. However, radiation enhances this inhibitory pathway by upregulation of CD47 in tumor cells, including breast cancer cells. Looking at both luminal (TSA) and triple negative (AT3) breast cancer models, the combination of radiation and anti-SIRPα was effective, although to different extents, which was ablated in the AT3 model by depletion of cDCs. As cDC1s do not express SIRPα, exactly which DC population was involved was puzzling. CITEseq of tumors after treatment revealed the presence of the expected cDC1 and cDC2 populations, but also a CCR7hi DC population with some markers of both cDC1s and cDC2s, and expressing CCL16 and SIRPα. These DCs are very similar to mregDCs identified by Miriam Merad. In vitro, these cells are very effective at dead cell uptake, and mechanistically, irradiation enhances phagocytosis by bone marrow-derived DCs, resulting in upregulation of CCR7. Antigen uptake is not impacted by the radiation-induced upregulation of CD47, suggesting the involvement of other “eat me” signals. Overall, Demaria sees great opportunities to use combinatorial strategies to optimize this simple and inexpensive approach to immunotherapy.
Neoadjuvant Combination Immunotherapy Approaches: Improving Outcomes and Propelling the Science - Thomas Marron, Icahn School of Medicine at Mount Sinai
To improve outcomes in metastatic cancers, Thomas Marron discussed the importance of research in the neoadjuvant setting. At this stage, more tissue can be obtained to inform eventual treatment development. His team has used window-of-opportunity (W-O-O) trials for this purpose, to answer questions about how agents work in patients and what underlies the heterogeneity of responses, thereby better predicting who will respond to therapy. Further, many drugs are in preclinical and clinical development, making it challenging to determine what to test, as there are limited patients available who meet the inclusion criteria to fill all open trials. With W-O-O trials, multiple therapies can be tested in a small cohort over a brief period (2-6 weeks), and tissue and blood samples can be collected before, during, and after the trial period, followed by in-depth analyses using single-cell technologies and methodologies to assess spatial interactions. For patients, this is beneficial, as the therapies are often effective due to the patient’s more intact immune system, smaller tumors, less heterogeneity, and less loss of heterozygosity. For research, it helps define mechanisms of action (MoA) and aids in detecting biomarkers, which can reduce the number of patients treated with ineffective treatments and helps design rational combinatorial approaches. In one such trial, the NeoCOAST study, patients received anti-PD-L1 alone or in combination with anti-CD73 or anti-NKG2A. The combinations were more effective in just 28 days, particularly the combination with anti-NKG2A. Using the collected samples to determine the MoA, the researchers uncovered anti-NKG2A-mediated upregulation of CXCL13 and the formation of tertiary lymphoid structures (TLSs). Another neoadjuvant study focused on hepatocellular carcinoma, with patients receiving 2 cycles of anti-PD-1 alone, or anti-PD-1 with prior stereotactic body radiation therapy (SBRT). After these treatments, patients underwent surgery and received adjuvant anti-PD-1. The neoadjuvant treatment improved outcomes, and a pathologic complete response was a good predictor of cure. Comparing the two study groups, there was little difference in pathologic response; however, disease-free survival was longer in the group receiving SBRT before anti-PD-1 therapy, potentially due to de novo T cell priming. Studying the tumor immune microenvironment (TIME) of the anti-PD-1-treated tumors, showed that many were T cell-rich, but only half of those were responders. Further investigation revealed that responders had high levels of effector T cells, as well as CXCL13+CD4+ Tfh-like cells, which were clonal and might be antigen-specific. Activated mregDCs localized with these CXCL13+ CD4+ T cells in the TIME. mregDCs are mature DCs that have taken up lots of antigen and upregulated immunoregulatory markers. While these cells were expected to limit T cell responses in the TME, they were present across immune-hot and -cold tumors. PD-1hi progenitor CD8+ T cells clustered with these mregDCs and with CXCL13+ Tfh cells; although progenitor cell numbers were similar across groups, they colocalized with mregDCs and Tfh only in responders, forming an immune triad. This suggested that mregDC were important for maintaining the CD8+ progenitor population, resulting in effector cells in response to anti-PD-1, in contrast to the formation of terminally exhausted CD8+ T cells in the T cell-rich non-responders upon anti-PD-1. Further assessing these triads and TLSs, Marron and colleagues found that responders exhibited plasma cell clonal expansion and had higher expanding and de novo tumor antigen-specific antibody responses. Therefore, these data suggest that TLSs might serve as biomarkers, and that identifying a therapy that can induce TLSs before surgery might improve response rates. Marron ended his talk by suggesting that efforts be made to maximize the use of collected patient materials in trials. To achieve this, he started an academic research organization (ARO) to facilitate collaboration with industry to enable these in-depth analyses.
Reading suggestion:
Personalized neoantigen vaccine improves anti-PD-1 in advanced hepatocellular carcinoma in early clinical trial
By Ute Burkhardt, Ed Fritsch, Maartje Wouters, and Lauren Hitchings
(This Keystone Symposia conference is available for On Demand viewing! Find out more here).



