Inhibition or reprogramming of tumor-associated macrophages (TAMs) has great potential to improve the tumor immune environment, but has, so far, shown limited clinical efficacy. To improve macrophage-targeted therapies, von Locquenghien, Zwicky, Cie, et al. described their dual TAM- and T cell-targeting strategy in a recent Cell publication.
Using publicly available datasets from human lung, breast, colon, and ovarian tumors, the researchers started by characterizing the spatial interactions between TAMs and other immune cells in the tumor microenvironment (TME). TAMs were the main population observed near T cells. Next, various algorithms were applied to a single-cell RNA sequencing atlas with human tumor samples and adjacent healthy tissues to assess cell–cell communication. This also showed that the predominant interaction was between TAMs and T cells, and involved chemokines related to T cell recruitment, costimulatory and inhibitory interactions, and immunosuppressive signals, which were all upregulated in the TME.
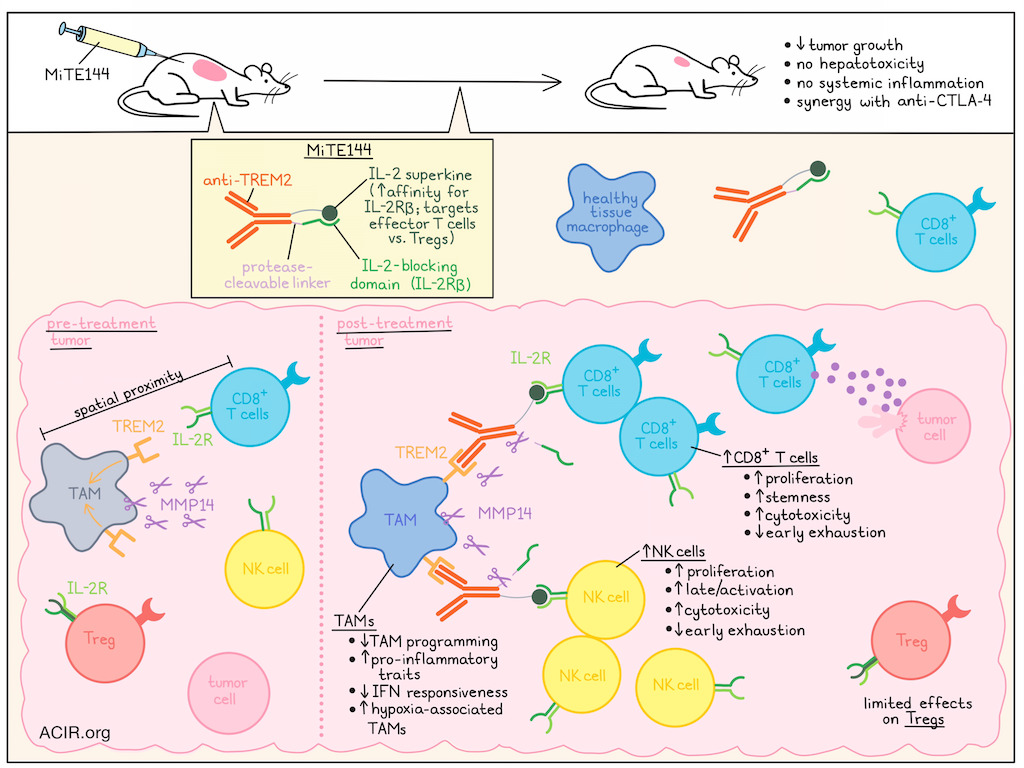
Based on these data, the researchers developed a treatment strategy to simultaneously reprogram TAMs and activate T cells in the TME. Previous work revealed TREM2 as an important inhibitory receptor on TAMs. Therefore, an antagonistic anti-human TREM2 antibody was developed and tested in humanized TREM2 mice. Treatment with this antibody prevented the induction of the TAM program, and preserved pro-inflammatory characteristics in vitro in bone-marrow-derived macrophages. However, the treatment had limited efficacy in mice with MC38 tumors. To improve efficacy, a myeloid-targeting immunocytokine (ICK) was created by fusing the anti-TREM2 antibody to IL-2, enabling it to act on both TAMs and T cells in the TME. A synthetic IL-2 variant (superkine [SK]) with increased affinity for IL-2Rβ was used, as it preferentially targets effector T cells over Tregs. However, intraperitoneal treatment of MC38-bearing mice with anti-TREM2-IL-2SK resulted in death, caused by systemic toxicity and hepatotoxicity.
To overcome this systemic toxicity, the researchers developed an inactive IL-2SK domain (pro-cytokine) containing a blocking domain (IL2-Rβ) with a protease-cleavable linker, which could be activated locally by TAM-associated proteases. To determine which protease to use for this construct, scRNAseq analysis of protease expression was performed on immune cells from a human cancer atlas, and PBMCs from healthy donors. TAMs showed higher expression of several proteases, and MMP14 was selected for its high expression in TAMs and limited expression in other immune cells. There was also a strong correlation between MMP14 and TREM2 expression in both human and murine tumors. Based on this, four variants of myeloid-targeted ICKs (MiTEs) were designed, and one of these was selected for further assessment. MiTE144 showed high affinity and specificity for TREM2, did not bind IL-2Rβ in its intact form, induced strong T cell proliferation after protease treatment, and had limited impact on Treg activity.
To determine its toxicity, mice inoculated with MC38 cells received MiTE144 intravenously every 3 days starting 10 days after tumor inoculation. No systemic inflammation or hepatotoxicity was observed. For efficacy analysis, mice were treated on days 8, 10, and 13 after MC38 tumor inoculation. MiTE144 monotherapy significantly reduced tumor growth and was more effective than anti-TREM2 monotherapy, immune checkpoint blockade (ICB) monotherapies, and combinations of anti-CTLA-4 and anti-PD-1 with anti-TREM2. The combination of MiTE144 with anti-CTLA-4 further improved its efficacy, with complete tumor eradication in 6/7 mice.
The researchers then analyzed the effects of MiTE144 on tumoral immune populations using scRNAseq on day 11 post-tumor inoculation, after mice received treatment on days 8 and 10. A deep generative model was used to construct treatment-specific distance similarity networks. Treatments with MiTE144, +/- anti-PD-1/CTLA-4, formed a distinct cluster. Differential expression analysis showed that therapies with MiTE144 triggered the most extensive transcriptional reprogramming in macrophages, DCs, T cells, and NK cells. It reduced IFN-responsive TAMs and DCs while increasing hypoxia-associated TAMs and granulocytes. In contrast, it increased CD45+ immune cell counts, monocytes, and granulocytes in TDLNs.
Using a deep learning model to assess the effect of each treatment relative to other treatments revealed that MiTE144 upregulated inflammatory monocyte-like and hypoxia-associated modules, while reducing IFN-response genes and modules. It also downregulated the suppressive hTREM2/Mmp14 modules in macrophages, while upregulating Trem1/3, suggesting a shift toward earlier, inflammatory monocytic states.
MiTE144 alone or combined with anti-PD-1/CTLA-4 increased NK cell proliferation, resulting in higher NK cell counts in the TME and TDLN. NK cells upregulated late/activation, cytotoxic, and proliferation genes, while early exhaustion and dysfunction-related genes were downregulated, suggestive of a shift toward a highly proliferative and cytotoxic phenotype.
Treg frequencies increased slightly in the TME and more in the TDLN, but this effect was not present when treatment was combined with anti-CTLA-4. CD8+ T cells increased in both the TME and TDLN in response to MiTE144, and these cells upregulated cytotoxic, proliferation, and stemness-associated genes, while exhaustion-associated genes were downregulated. These effects were more pronounced when MiTE was combined with anti-CTLA-4.
To determine whether these effects could also be observed in human tumors, patient-derived tumor fragments (PDTFs) obtained from five patients with renal cell carcinoma were treated ex vivo with MiTE144 and/or anti-PD-1, and immune cells were analyzed by CITEseq after 48 hours. One non-responding PDTF was excluded from analysis. In the other PDTFs, MiTE144 +/- anti-PD-1 resulted in shifts in immune cell populations, with expansion of CD8+ cycling and memory T cells and intermediate-stage NK cells. Tregs and hypoxic TAMs remained unchanged or reduced compared to controls. T cells upregulated cytolytic effector genes and cell cycle-associated genes, while markers associated with exhaustion decreased. In NK cells, treatment also induced cytotoxic programs, proliferation-associated genes, and activation markers, while downregulating exhaustion genes.
Overall, this MiTE strategy enabled reprogramming of TAMs, improved T cell activity, and worked synergistically with ICB. If shown to be effective and safe in patients, it may serve as an interesting new combinatory strategy with ICB, particularly for tumor types with high numbers of immunosuppressive TAMs that currently show limited response to ICB treatment.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first author Michelle von Locquenghien answered our questions.

What was the most surprising finding of this study for you?
The most unexpected result emerged during the first generation of anti-TREM2–IL-2 constructs. We initially assumed that the TREM2-binding Fab would provide sufficient tumor selectivity to prevent off-target cytokine activity. Instead, our first in vivo studies in transgenic hTREM2 mice resulted in acute cytokine-driven systemic toxicity, revealing how deceptively small design assumptions can have major biological consequences. These results prompted a complete reassessment of our engineering strategy, and reshaped our underlying hypothesis. In parallel, while we anticipated strong T cell activation and proliferation in response to the IL-2 stimuli, the magnitude and consistency of NK cell cytotoxicity and proliferation across models were far greater than expected. This synergistic myeloid–lymphoid response may represent a critical advantage for overcoming challenges that limit T cell-centric immunotherapies, such as MHC-I loss.
What is the outlook?
This work positions MiTEs as a next-generation macrophage-targeted immunocytokine platform that integrates conditional cytokine activation with immune checkpoint blockade. Because MiTEs function independently of tumor antigens and become active specifically within the tumor microenvironment, they offer broad applicability across solid tumors while avoiding systemic toxicity, overcoming key limitations of traditional cytokine therapies. MiTEs show promising results in human patient-derived tumor fragments and synergize strongly with existing immune checkpoint inhibitors. The fully humanized design supports a clear translational path, including non-human primate studies and eventual clinical evaluation. Beyond IL-2, the platform can be extended to additional cytokines and immune axes to overcome resistance mechanisms and drive multi-dimensional immune reprogramming in cancer therapy.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
View unexpected results as strategic opportunities rather than setbacks; some of the most impactful insights arise when data challenge your assumptions. And be willing to pursue ambitious, high-risk ideas. Transformative research often begins with daring to explore concepts that push past traditional approaches.



