
The ACIR team attended the SITC - 40th Anniversary Annual Meeting 2025 in National Harbor, Maryland. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Keynote address
Jennifer A. Wargo
Richard V. Smalley Memorial Award
Ira Mellman, PhD
Epitopes and cancer vaccines
Catherine J. Wu
Christopher Klebanoff
Olivera Finn
Dustin McCurry
Rom S. Leidner
Tumor immune microenvironment
Florent Ginhoux
Jennifer L. Guerriero
Jianjun Zhang
Immune checkpoint blockade
Moran Amit
Bertrand Routy
Jake Lichterman
Aurélien Marabelle
T cell and NK cell therapies
Julian Lum
Haolong Li
Beyond
Adam Grippin
Margaret K. Callahan
Allen S. Yang
Keynote Address
Targeting the Microbiome to Promote Health and End Cancer- Jennifer A. Wargo, MD, MMSc – The University of Texas MD Anderson Cancer Center
Jennifer Wargo delivered a comprehensive keynote lecture on the discovery of the link between microbes and the immune system and the opportunities that the ecosystem has generated to improve outcomes for patients with cancer. Microbes are pervasive, and the microbiome's genetic material within a person dwarfs that of human genes, with 100–1000 times as many microbial genes compared to human genes, and polymorphic microbes now being a hallmark of cancer. Wargo’s study of the tumor microbiome began serendipitously when a stromal cell line that appeared to confer broad resistance to gemcitabine was found to be contaminated with Mycoplasma. Importantly, the team persisted, showed that the mycoplasma were breaking down the drug, and went on to validate this in human pancreatic cancer and in mouse models, where they found that intratumoral Gammaproteobacteria similarly conferred resistance to gemcitabine. Microbial signatures have now been found across all tumors, but such analysis can be complicated and prone to artifacts. To ensure rigor in microbiome studies, Wargo and others have created the Platform for Innovative Microbiome and Translational Research (PRIME-TR). While intratumoral microbes can promote cancer, they also offer new targets and potentially can mediate antitumor immunity via microbial neoantigens. In addition, gut microbes, with a long trail of interaction – and education – between luminal microbes and the network of immune cells below the endothelial cell layer, can also profoundly impact immunity and immunotherapy response. Seminal work showed that high gut microbial diversity correlates with better survival after stem cell transplant in patients with leukemia, and that modulating the gut microbiota could lead to better responses to immune checkpoint blockade (ICB) in mouse models, and then in multiple studies in patients with melanoma. Specific microbial signatures (and probably the absence of deleterious taxa) also show promise as markers of response to ICB or CAR T cell therapy. In 2021, two clinical studies showed that modulation of the gut microbiome by fecal microbiota transplantation (FMT) from responding patients enhanced responses to immunotherapy. Building on, later work showed that FMT from healthy donors could also improve outcomes, but recognizing that it was difficult to understand which healthy donors were useful, Wargo and her team engineered a next-generation synthetic microbial community based on a complete responder donor's material. This engineered consortium augmented ICB response as well as the whole FMT in preclinical models. Turning to probiotics, although an initial paper suggested live bacteria were beneficial, retrospective analysis of a set of patients by Wargo and colleagues hinted that patients taking over-the-counter available probiotics had worse outcomes, which was demonstrated in a preclinical model. Coupled with a well publicized case study, these results led Wargo and colleagues to generally recommend patients against the use of probiotics. Antibiotics, diet and environmental factors can all influence gut microbes and immunotherapy responses, but there are nuances. Pre-ICB antibiotics typically worsen outcomes, though some targeted antibiotics may enhance responses. Based on dietary and lifestyle surveys, patients who reported sufficient dietary fiber intake of ≥20 grams/day were five times more likely to respond to ICB, which was confirmed in mice. In addition, dietary fiber can shift the TME via monocyte reprogramming. In a randomized controlled feeding study in patients with resectable metastatic melanoma on ICB showed that a fiber-rich diet (up to 50 grams/day) nearly tripled the response rate and doubled event-free survival compared to a 20 grams fiber/day diet. Post-high-fiber-diet fecal microbiomes conferred better tumor control in germ-free mice than pre-diet microbiomes. Next, studying over 150 gliomas and metastatic brain tumors, Wargo and her team found distinct oral bacteria like Fusobacterium in these tumors, suggesting that poor oral hygiene may allow these microbes to translocate and associate with disease progression. Wargo closed with a concerned look at the increasing rates of inflammatory disease and cancer rates among younger individuals, possibly related to overuse of antibiotics and poor diet, and a plea for multi-pronged changes that positively impact an individual person’s health (with knock-on effects to our planet's health).
Reading suggestion:
New gut-associated checkpoint might be involved in ICB resistance in cancer.
Location, location, location: why lung tumors trigger weak immune responses in mice.
Richard V. Smalley Memorial Award
The Coming Renaissance of Cancer Immunotherapy- Ira Mellman, PhD – Parker Institute for Cancer Immunotherapy, Medici Therapeutics
In honor of his outstanding career as an academician and in industry, and the high impact of his work in basic immunology and cancer immunotherapy, Ira Mellman received SITC’s most prestigious award, the Richard V. Smalley Memorial Award. Mellman began by describing how conceptualization of the cancer immunity cycle was inspired by the desire to simplify the emerging complexity of immunology and identify the rate-limiting steps, in order to focus on the approaches to overcome them. Two key parts of that cycle – stimulating T cell activation, and keeping T cells active in the tumor – have been the basis of the first, and still largest successes in immunotherapy: immune checkpoint blockade (ICB). Despite the success of ICB and other immunotherapies over the past decade (T cell engagers, CAR T cells, etc.), a next big step has not emerged for solid tumors. Anti-PD-(L)1 therapies have been the greatest success, but they emerged not from a highly focused, mechanistic investigation by industry, but from an academic-inspired idea and a small company willing to give it a try. The community is only now recognizing that the mechanism of action is much more complicated than initially considered. We now appreciate an outsized role for the PD-1 pathway in abrogating costimulation, not just TCR stimulation, and that PD-L1-expressing myeloid cells (particularly dendritic cells), not tumor cells, have the biggest role. These complexities may also help explain why anti-TIGIT therapies have not yet demonstrated their anticipated success. Inhibition of the negative signals from TIGIT may be redundant with those from anti-PD-(L)1, essentially “doubling down” on an already impacted mechanism, with the added anti-TIGIT effect being insufficient to further improve efficacy. Mellman pointed out that the emerging focus on understanding the immunosuppressive TME may ultimately identify additional rate-limiting steps that could better capitalize on the different, but overlapping mechanisms. Reflecting on his experience, Mellman described that going forward, he is most excited to focus on two modalities that show efficacy or strong hopes for efficacy in the clinic, but that are still underexplored at the research and development end – cell therapies for solid tumors (as evidenced by the clear success of TIL therapy) and cancer vaccines. The collaboration between Genentech and BioNTech on personalized neoantigen cancer vaccines in pancreatic cancer provided clear evidence in the adjuvant setting that immune response correlated with improved relapse-free survival and evidence for immunosurveillance, resulting in clonal evolution. However, a study in more disease types in the metastatic setting revealed that a strong immune response was insufficient to generate tumor control, and that maintaining a memory effector T cell population was critical. Important remaining vaccine questions include, among others, better adjuvants, better delivery approaches, and a better understanding of the role of CD4+ T cells. As an example of the latter, Mellman described recent experiments in which a single MHC class II-restricted (and non-MHC class I) epitope derived from an expressed endogenous retrovirus (ERV) led to tumor control, which depended on CD8+ T cells, the antigen specificity of which are under investigation. Finally, he closed by discussing the demonstrated examples of TIL therapy leading to cures, and the exciting emerging potential to use advanced structural modeling to predict TCR structures to generate synthetic TCR “TIL” libraries in silico on a patient-specific basis and in vivo nucleic acid delivery to create an inexpensive, personalized alternative to TIL therapy, overcoming a major limitation.
Reading suggestion:
Novel binder designs mimic TCR specificity for pMCH-I.
First signs of clinical efficacy for personalized TCR-engineered T cells.
Epitopes and cancer vaccines
The Dark Genome as a Source of Candidate Tumor Antigens- Catherine J. Wu, MD – Dana Farber Cancer Institute
Immunotherapy, in all its different forms, is critically dependent on tumor antigens (the "sharp point of the spear"), and aims to expand existing T cells and generate new responses against these antigens. Cathy Wu and others have initiated a resurgence of cancer vaccines by using next-generation sequencing to identify somatic mutations that generate new tumor vaccine targets – neoantigens – and enable personalized vaccination, which has proven safe, feasible, and immunogenic, with evidence of on-tumor targeting and reduced recurrence in adjuvant settings across various cancers. However, the scarcity of high-quality somatic targets poses a challenge in the majority of tumors with low mutation burdens. While targeting tumor-associated antigens, cancer-associated viruses and somatic mutations represent only the tip of the iceberg. Exploring the "dark proteome” – cryptic antigens, mistranslated products, or hypoxia-induced endogenous retrovirus (ERV)-derived neoantigens – may provide more opportunities for further effective antigen discovery. Ribosequencing demonstrated active translation across previously unannotated open reading frames (nuORFs), and immunopeptidome data confirmed that these nuORFs contributed to class I HLA-bound peptides. There are many species of nuORFs defined by their origin (linc RNAs, 5’ UTR, etc.) and many constitute neoantigens by harboring cancer-specific mutations or by cancer-specific transcription and translation. Encouragingly, T cells carrying TCRs with specificity for those cryptic antigens demonstrated robust cytotoxicity in vitro, and inhibition of tumor growth in vivo. Wu then turned to novel antigens derived from hypoxia-induced ERVs. Normally silenced, ERVs occupy up to 8% of the human genome, and can be reactivated in tumors. Historical data in renal cell carcinoma (RCC) showed an ERV-derived peptide recognized by T cells, and this RCC-specific ERV upregulation was driven by HIF-2. Confirmation studies showed ERV translation is HIF-2-responsive and -regulated. Using class I ligandome analysis, immunogenic ERV-derived peptides could be identified in patients. Going back to the historic clinical data, anti-ERV reactivity correlated with clinical responses in RCC patients undergoing allotranslation, suggesting that in the future, clinical-grade HIF-stabilizers could also activate this target class in non-RCC tumors. Next, Wu set out to enhance the discovery of alternative tumor-specific antigens with mass spectrometry (MS). While traditional MS lacks sensitivity for low-abundance targets, data-independent acquisition (DIA) MS is more sensitive, but requires a spectral library of searched peptides as a reference. Wu and her team developed Pepyrus, which uses E. coli to create large, user-defined peptide libraries and reference spectra. Pepyrus-DIA is significantly more sensitive than conventional methods, successfully detecting previously undetectable neoantigens, nuORFs, and ERV antigens in melanoma and RCC patient samples. The reference library proved reusable ("off-the-shelf") across different cancers. This enhanced antigen target discovery is promising, but clinical translation requires answering critical questions about T cell killing efficiency, TCR characteristics (affinity, functionality), and de-risking for off-target toxicity.
Reading suggestion:
Neoantigen vaccination shows promising prevention of recurrence of high-risk kidney cancer.
Hidden gems: cryptic antigens as novel therapeutic targets in pancreatic cancer.
Dark Proteome Using TCR Gene Therapy: RNA mis-splicing derived public neoantigens- Christopher Klebanoff, MD – Memorial Sloan Kettering Cancer Center
Christopher Klebanoff presented work on targeting cryptic neoantigens that arise from recurrent RNA mis-splicing events, a common feature of cancers with splicing factor (SF) mutations. SF mutations are prevalent in cancers, including myeloid malignancies and solid tumors like lung and pancreatic cancers, and are associated with poor prognosis. Since SF mutations can cause mRNA sequence changes, alter the cancer cell proteome, and are recurrent across patients’, Klebanoff and team hypothesized that these early driver mutations create an elite class of shared, clonally conserved, and – because these events can create long novel open reading frames – numerous neoantigen opportunities. Comprehensive analysis of alternative splice forms in SRSF2-mutant (SRSF2p95) myeloid malignancies identified 56 HLA-A2-restricted candidate neopeptides, predicted using NetMHC, that were absent in SRSF2WT myeloid cancers and in normal tissues. A T2 peptide stabilization assay confirmed that approximately two-thirds of the predicted neopeptides were stable binders. One promising SRSF2p95-derived candidate resulted from an Exon 4-skipping event in the CLK3 gene. Immuno-peptidomic studies on SRSF2p95/WT isogenic K562 cells identified the predicted CLK3 neopeptide on the cell surface presented by HLA-A2, confirming endogenous processing and presentation. To assess human immunogenicity, biospecimens from HLA-A2+, SRSF2-mutant patients were probed with dextramers loaded with the neopeptides, resulting in the identification of neoantigen-reactive CD8+ T cells. scRNA/TCR sequencing was subsequently used to nominate candidate TCRs. Neoantigen-reactive and viral-reactive CD8+ T cells showed a terminally differentiated phenotype, but neoantigen-reactive CD8+ T cells had lower expression of genes associated with TCR signaling and effector function compared to virus-specific cells, consistent with other neoantigen studies. As an example, isolation of one paired TCR (TCR-2) from patient-derived T cells and re-insertion into polyclonal T cells triggered IFNγ and TNFα production when co-cultured with target cells electroporated with the mis-spliced CLK3 mRNA, but not the wild-type form. This activation was co-receptor independent (i.e., functional in both CD8+ and CD4+ T cells), a feature associated with high-quality TCRs. Furthermore, TCR T cells co-cultured with primary HLA-A2+ SRSF2-mutant tumor cell lines, representing physiological presentation, demonstrated significant activation against the SRSF2-mutant, but not healthy donor cells. A therapeutic proof of concept was shown in a luciferase-expressing xenograft model, where the CLK3-TCR T cells conferred durable disease control. Klebanoff also highlighted that allogeneic transplantation in MDS-EB1 patients induced large clonal expansions of terminally differentiated T cells; many of these T cells were found to recognize mis-spliced neopeptides and were co-receptor-independent. The approach is generalizable to solid tumors, as Klebanoff also identified an immunogenic mis-spliced neoantigen from the GNS gene in glioblastoma multiforme. Klebanoff concluded that SF mutations are prevalent, and generate a rich pool of actionable and shared neoantigens, and that novel TCR gene therapy is a promising strategy for both hematologic and solid tumors.
Reading suggestion:
Tumoral RNA splicing: source of neoantigens for off-the-shelf therapies?
MUC1 Vaccine Targets Pre-Cancer: The Path to Primary Prevention- Olivera Finn, PhD – University of Pittsburgh
Olivera J. Finn presented her work on cancer vaccines targeting MUC1, a tumor cell surface antigen that is overexpressed and abnormally and severely under-glycosylated in over 80% of human tumors, making its altered form tumor-specific. MUC1 is crucial for tumor progression, and cannot be lost by tumor cells. Preclinical data showed that a synthetic non-glycosylated MUC1 peptide vaccine plus poly-ICLC adjuvant successfully induced specific antibodies and T cells (which did not react to normal tissue MUC1), and led to tumor rejection upon intradermal or intramuscular injection. Past therapeutic trials (dating back to 1993-2010) in advanced cancers led to measurable immune responses and prolonged time to recurrences in only about 20% of the patients, and this limited activity was eventually attributed to the highly immunosuppressive tumor microenvironment. Other vaccines at the time faced a similar challenge. This led to a shift toward cancer interception and prevention, targeting MUC1 when it appears in premalignant lesions, before severe immunosuppression develops. In a clinical trial, individuals with a history of premalignant colorectal adenomas (polyps) were vaccinated after surgery. Compared to patients with advanced cancers, vaccinated patients showed significantly higher (30–40x) anti-MUC1 IgG responses, and immunological responses were observed in a significantly higher fraction of patients (about 50% vs about 2-3%). In the patients who did not respond to the vaccine, Finn observed higher levels of circulating myeloid-derived suppressor cells (PMN-MDSCs). In a randomized, double-blind phase 2 trial, new polyp recurrence at 3 years was only 27% in vaccine responders, compared to 64% and 66% in non-responders and the placebo group, demonstrating a strong efficacy signal. In an ongoing trial in ductal carcinoma in situ (DCIS), a MUC-1-expressing precursor to breast cancer, the vaccine showed high immunogenicity, eliciting a more than 2-fold increase in serum anti-MUC1 IgG in most patients. Finn summarized that vaccinating in premalignant and high-risk settings is feasible and generates high-level immune responses and long-term memory, She also advocated for repurposing other tumor antigen vaccines for interception to treat individuals at high risk for cancer.
Reading suggestion:
Non-mutated tumor antigens in the spotlight for therapeutic targeting.
Immune escape through oncogene-mediated editing of the HLA-ligandome- Dustin McCurry, MD – The University of Texas MD Anderson Cancer Center
Dustin McCurry outlined efforts to identify novel oncogene-driven mechanisms of immune escape in well annotated cohorts of patients with myeloid malignancies treated with Donor Lymphocyte Infusion (DLI) and followed over time. DLI responders were characterized by terminally exhausted T cells pre-DLI, and precursor exhausted T cells post-DLI; no clear pattern emerged in non-responders, leaving open the cause of DLI resistance. Hypothesizing that novel genomic drivers confer resistance to DLI, McCurry performed genomic and scRNA sequencing on a cohort of 21 patients. Whole-exome sequencing did not identify known genomic drivers in responders beyond BCR-ABL. In contrast, non-responders showed multiple mutations, with ASXL1 being the most frequent in 3 of the 4 patients who didn’t respond. ASXL1 is a common mutation in clonal hematopoiesis and myeloid malignancies, and is associated with poor prognosis. ASXL1 is associated with dysregulated histone modification through interaction with polycomb proteins, but has never been linked to an immuno-evasive role. An extended cohort confirmed that ASXL1 was the most frequent mutation in non-responders (n=9), and was absent in responders. To determine how ASXL1 mutations drive DLI resistance, the McCurry and team analyzed scRNAseq data and identified four cell states enriched in non-responders.These states were all linked to hematopoietic cell precursor states, in particular, associated with hematopoietic stem cells and enriched in leukemic stem cell signatures. Mechanistically, HLA-II expression was unchanged; however, non-responders showed a significant decrease in HLA-I expression. To functionally validate the class-I involvement, the researchers used K562 cells, which both endogenously lack HLA-I expression and carry an ASXL1 mutation. CRISPR-mediated correction of the mutant allele induced baseline HLA-I expression, which was strongly potentiated by IFNγ. Conversely, in the HLA-I positive AML cell line U937, introduction of the ASXL1 mutation suppressed HLA-I expression. In a T cell killing assay, engineered T cells against K562 (a specific antigen) did not kill the mutant K562 cells, but were able to kill the ASXL1-corrected (HLA-I re-expressing) cells. In addition, scRNAseq data also revealed differences in antigen presentation machinery, specifically immunoproteasome genes. Mass spectrometry-based immunopeptidomics revealed very few unique peptides on the ASXL1 mutant cell line, but an increase in the corrected cell line. To determine if this was due to HLA-I quantity, both cell lines were engineered to express equal levels of HLA-A*02:01. Even with matched A2 expression, the ASXL1-corrected line presented significantly increased peptide diversity. Furthermore, the source genes for differentially presented peptides were not differentially expressed, suggesting the ASXL1 mutation decouples the peptide diversity from the cellular transcriptional state. Analysis of the A2-presented peptides revealed differential amino acid usage at key anchor residues (P2 and P9) in both corrected and mutant cell lines, with altered peptide cleavage motifs in the ASXL-mutant cell line. This was clearly exemplified by the housekeeping gene GAPDH, which presented completely different, unique peptides in the mutant versus the corrected line. This remodeling was shown to reduce immunogenicity, as the ASXL1-mutant cells presented significantly fewer strong-binding peptides and restricted the immunoproteasome. McCurry concluded that ASXL1 is a novel immune escape-associated oncogene that drives DLI resistance via epigenetic suppression of HLA-I expression, restricting the presentation of high-affinity peptide epitopes.
Proteasomal Cargo Biologic in Prime/Boost with PD-1 Blockade- Rom S. Leidner, MD – Earle A. Chiles Research Institute, Providence Cancer Institute
In contrast to an in silico or immunopeptidome-directed vaccine strategy, Rom Leidner pursued the idea of a vaccine comprised of the undefined mixture of short-lived proteins (SLiPs) created by shuttling of cell proteins to the autophagosomal pathway following blockade of the proteasome, as pioneered by Hong-Ming Hu. Two lung tumor-derived cell lines (one from a patient with squamous carcinoma and one from a patient with adenocarcinoma) were used to prepare DPV-001, an autophagosomal/proteasomal cargo vaccine, for a Phase I clinical trial. The vaccine consists of spectrin- and F-actin-coated double-membrane microvesicles, which target CLEC9A dendritic cells, and contain multiple damage-associated molecular patterns (DAMPs), toll-like receptor (TLR) ligands, the associated partially degraded SLiPs, defective ribosomal products (DRiPs), tumor-associated antigens (TAAs), and over 400 non-canonical peptides. Immunopeptidomics identified that these “dark matter” antigens derived mostly from 5’UTR non-coding RNAs and off-frame translation. Key highlights from the preclinical work were that the vaccine showed synergy with a GITR agonist and anti-PD-1, and that the combination of anti-PD-1 and OX40 agonism was effective only when agOX40 preceded (during priming) anti-PD-1, since PD-1 is necessary to enhance immune memory. The Phase I study focused on advanced HNSCC, and the design included 72 hr pretreatment with cyclophosphamide, DPV-001 vaccine with or without GITR agonism, and two weeks later, anti-PD-1. Eleven doses of the vaccine were delivered over a year, and treatment continued for two years. Toxicity was tolerable and all immune-related, so considered a “good sign”. Eight of 18 patients had a clinical response, with the GITR arm outperforming the control arm, and the overall and progression-free survival exceeding historical controls of anti-PD-1 alone. Some patients with multiple poor prognostic markers (prior anti-PD-1 therapy failures, low PD-L1 CPS) and multiple metastatic lung nodules nevertheless responded. Immunologically, an early increase in proliferating CD4+ effector memory cells was observed, and later included CD8+ effector memory. In tumor biopsies, stronger infiltration of CD8+ memory and effector memory cells was observed in responders than in non-responders. Determining antigen specificity of this “hyper-polyvalent vaccine” is a challenge, but analysis of expanded TCR clonotypes showed reactivity to autologous and HLA-matched or partially matched cells, even from other histologies, suggesting shared targets. Notably, responses were not detected prior to vaccination, suggesting only transient, non-immunogenic presentation – a frequent observation for dark matter epitopes. Finally, analysis of non-responders suggested an increase in LAG-3+ T cells, supporting a further trial including anti-LAG-3 treatment.
Reading suggestion:
Hydrogelation preserves whole tumor cells for vaccination.
Tumor immune microenvironment
Harnessing Tumor Associated Macrophage Heterogeneity- Florent Ginhoux, PhD – Gustave Roussy
Florent Ginhoux presented work on the heterogeneity of tumor-associated macrophages (TAMs), arguing that the M1/M2 paradigm is a non-productive oversimplification for the field. TAMs co-opt normal, everyday macrophage functions to support tumor progression, and a deeper understanding based on ontogeny, spatial location, and activation state is required to understand the spectrum of macrophage states within the tumor. Adult macrophages are either resident tissue macrophages (RTMs) of embryonic or early developmental origin, or are derived from adult bone-marrow monocytes, which can be recruited to tumors. To track the monocyte-derived macrophages the researchers used an Ms4a3-Cre-dtTomato fate-mapping mouse model (dtTomato+ cells are monocyte-derived) bearing orthotopic pancreatic (KPC) tumors. Ginhoux’s team found that in established tumors, about 20-30% of TAMs were not monocyte-derived, but were persistent, pre-existing RTMs; these RTMs were also found in lung and breast tumors. Bulk RNA sequencing of these sorted populations revealed they were strikingly different transcriptionally. The Tomato+ monocyte-derived TAMs expressed genes associated with T cell activation and cytokine production, while the Tomato- embryonic RTMs were linked with homeostatic macrophage functions, such as matrix remodeling and collagen catabolism. RTMs express CD206 (Mrc1) at high levels, and were present in the healthy pancreas before tumor implantation. To track RTMs in the tumor, the researchers generated dtTomato-labeled RTMs 7 days prior to tumor implant, and found that RTMs were restricted to the tumor periphery and the healthy pancreas, while the later-arriving monocyte-derived TAMs inhabited the tumor core. This ring of CD206+ macrophages at the tumor-invasive front was also confirmed in human PDAC samples. To define the role of peripheral RTMs, an Mrc1-Cre-DTR model was used to deplete them. Depleting RTMs at an early stage (but not at a late stage) reduced tumor progression and epithelial-mesenchymal transition (EMT) in the cancer cells. Spatial analysis confirmed that the RTMs were preferentially co-localized to tumor regions exhibiting a high EMT signature at the periphery, suggesting a role of RTMs in promoting EMT. To determine the mechanism, the researchers used a slow-progressing autochthonous pancreatic intraepithelial neoplasia PDA model, and found that RTMs were the dominant macrophage type present during early-stage metaplasia, and that the EMT signature accumulated over time in correlation with RTM presence. A screen for RTM-specific secreted factors identified selenoprotein (SELENOP; SEPP1), a key selenium transporter. SELENOP was highly expressed by the CD206+ RTMs, and genetic deletion of SELENOP in RTMs led to slower tumor growth, improved survival, and reduced EMT in the KPC model. Spatial analysis showed that the tumor core was selenium-deprived, whereas selenium accumulated at the invasive front of the tumor. Furthermore, cancer cells showed an inverse correlation between SELENOP expression and EMT, and a positive correlation between ferroptosis and EMT. Ginhoux hypothesized that the RTMs at the periphery secrete selenoprotein, which transfers selenium to the adjacent cancer cells. This selenium is a crucial cofactor for the enzyme GPX4, which functions to suppress ferroptosis and enable cancer cells to survive the EMT process. Selenoprotein-dependent transfer was confirmed in a co-culture experiment in which macrophages labeled with the selenium isotope were shown to transfer it to KPC tumor cells, which was rescued in SELENOP-knockout macrophages. Human PDAC tumors also showed decreases in spatial selenium density from the adjacent pancreas to the tumor. Ginhoux concluded that RTMs protect tumor cells from ferroptosis and promote EMT via selenoprotein, and that RTM-derived selenoprotein is crucial in EMT-mediated tumor progression in human patients.
Reading suggestion:
TAMing immunotherapy resistance by targeting TIM3+VISTA+ macrophages.
From Complexity to Therapy: Targeting Tumor-Promoting Lipid Macrophages with CAR-Monocytes- Jennifer L. Guerriero, PhD – Brigham and Women’s Hospital
Jennifer Guerriero presented a novel, first-in-class, strategy to eliminate suppressive lipid-associated macrophages (LAMs) using engineered CAR-monocytes. To set the stage, Guerriero highlighted that macrophages are abundant in most solid tumors, co-opt homeostatic wound-repair functions to promote tumor progression in many ways, and are correlated with worse relapse-free and overall survival. She argued that current macrophage-targeting strategies have not been successful yet, because they fail to account for the vast complexity of tumor-associated macrophage (TAM) subsets, and it is often unknown what subsets are being, or should be, targeted. Further, she strongly cautioned against using the M1/M2 nomenclature, stating it has slowed the progress in the field. To systematically unwrap this complexity, Guerriero’s team performed extensive single-cell transcriptional and matched tissue spatial multiplex Immunofluorescence analysis on breast tumors, using both an in-house Dana-Farber/Brigham cohort and publicly available datasets. Differential gene expression, cell correlation analysis, gene set variation analysis, and spatial location identified ten distinct TAM subsets. The two most frequent and suppressive populations identified were GPNMB+ TAMs and SPP1+ TAMs. Both subsets were associated with lipid metabolism, while SPP1+ TAMs also showed a hypoxic signature. The lipid phenotype was interesting, as Guerriero and her team had previously described a lipid-metabolic macrophage phenotype, characterized by high CD36, CSF1R, and TREM2 expression, that is induced by and confers resistance to PARP inhibitors (PARPi) in a BRCA-mutant TNBC mouse model treated with olaparib. Combining olaparib with an anti-CSF1R agent reduced suppressive TAMs and enhanced PARPi therapy, doubling median overall survival and remodeling the TME. This finding has been directly translated to the clinic in the now half-enrolled AXALAP Phase 1b trial, which combines olaparib with the anti-CSF1R antibody axatilimab. Guerriero also highlighted work from Christine Brown's lab showing that SPP1+ TAMs, which also express TREM2 and APOE, correlate with CAR T cell resistance in glioma patients. TREM2 was also identified in their single-cell data, was uniquely expressed in tumor macrophages, and was associated with poor clinical outcomes. An anti-TREM2 antibody has shown promising efficacy in preclinical models and is reported to be safe in phase 1 clinical trial. Finally, Guerriero introduced a first-in-class strategy – macrophage replacement – using CAR-monocytes to target and eliminate TREM2+ tumor macrophages, and leveraging the innate ability of monocytes to infiltrate and survive in the harsh conditions of solid tumors. Proof-of-principle data showed that both murine and human anti-TREM2 CAR-monocytes efficiently phagocytosed TREM2+ macrophages in vitro. In the in vivo EO771 breast cancer model, a twice-weekly infusion of a million CAR-monocytes resulted in a 50% reduction in tumor burden at day 14, and a significant survival benefit, with 100% of treated mice alive at day 20, when all control mice had succumbed. Guerriero concluded that these CAR-monocytes can travel to the tumor, eliminate suppressive TREM2+ TAMs, and can be further engineered to induce a durable, endogenous T-cell response across solid cancers.
Reading suggestion:
Focus on suppressive macrophages may boost effects of PARP inhibitors.
Reprogramming the Immune Microenvironment to Intercept Lung Adenocarcinoma Pre-cancer- Jianjun Zhang, MD, PhD – The University of Texas MD Anderson Cancer Center
Despite improvements in overall survival over the past decade, lung cancer is still a deadly disease with a poor outlook once diagnosed. Recognizing that earlier, local disease has better prospects, Jianjun Zhang built the case for an interception approach during the very early pre-cancer developmental stage to prevent the cancer from forming, reducing significant morbidity and mortality if successful. Better and more routine imaging techniques (not only for possible lung disease, but possible heart disease, etc.) have helped to identify early lung nodules (ground glass opascities), which can be followed and biopsied, providing a better map of the stages of lung adenocarcinoma development. Multiple stages have been identified in the progression from normal tissue to invasive disease, and this timeline extends for more than a decade, providing significant time for iterative therapies. Key challenges, though, include the complications of invasive biopsies, the indeterminate nature of many lesions when biopsied, and multi-focal disease, even at the pre-cancer stage, eliminating surgery or radiation as feasible options. Genomic analysis has revealed a gradual increase in cancer-supporting mutations, copy number variations, and methylation alterations, and immunological analysis has identified a weakening of the cancer-fighting immune system, suggesting that the best opportunity may be at the earliest stage – “weaker enemy, stronger army”. Zhang first described the IMPRINT trial, which delivered four doses of anti-PD-1 every 3 weeks to patients with indeterminate persistent lung nodules, and who met strict predictive criteria (the Brock University Score) for high-risk pre-cancer rather than patients with early-stage cancer. Blocking the PD-1/PD-L1 axis was chosen based on the observations that anti-PD-1 therapy is more effective when PD-L1 expression is high, and that PD-L1 is already upregulated early in lung pre-cancer growth, and increases with development stages. No major toxicities were seen (in fact, toxicities in these healthy individuals seemed milder than in patients with cancer) and nearly half of the patients showed some nodule shrinkage (up to about 30% shrinkage). Zhang then described a second trial, Can-Prevent-Lung, which used the anti-IL-1β antibody canakinumab, based on the roles that macrophage and IL-1β play in supporting the early inflammatory environment, and on observations from the very large, multi-year heart attack and stroke prevention trial with canakinumab, which revealed a decrease in lung cancer incidence. Individuals with multiple persistent nodules were treated for 6 months, and then followed for 2 years for safety. About two-thirds of patients responded with shrinking nodules (up to about 50% shrinkage) and with lower “energy” (fewer cells per lesion). Key challenges moving forward include developing better risk projections, better biomarkers to follow disease progression/regression and, importantly, new therapeutic strategies based on deeper biological and immunological analysis.
Reading suggestion:
Cancer escapes before you know it’s there.
Immune checkpoint blockade
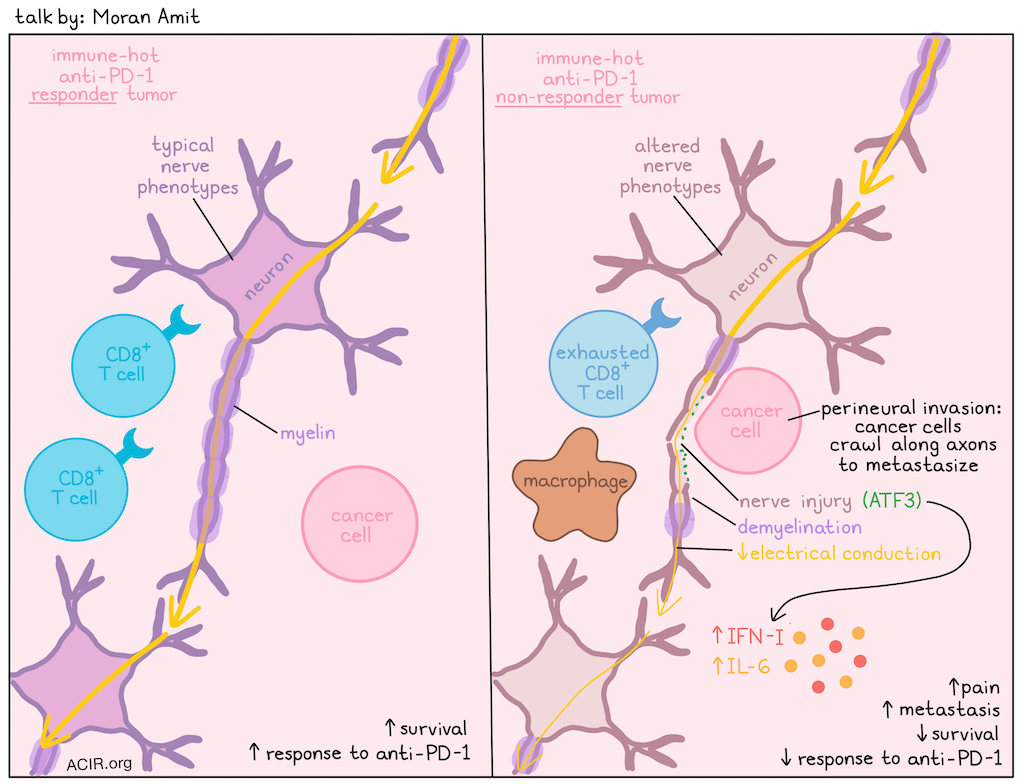
Nervous Tumors: Tumor-associated nerves promote resistance to immunotherapy- Moran Amit, MD, PhD – The University of Texas MD Anderson Cancer Center
Moran Amit explored the emerging field of cancer neuroscience, focusing on the critical role of nerves in tumor progression and immunotherapy resistance. While the presence of nerves in tumors has been known for more than 120 years, more recent research shows tumors transition neural signal dependence from sympathetic (adrenergic) to parasympathetic (cholinergic), and actively recruit nerves. Tumors possess a unique neural landscape, and the nerves within them change phenotype (e.g., sensory nerves acquiring adrenergic features), affecting clinical outcomes like pain, metastasis, and survival. Furthermore, there is a crosstalk between nerves, tumors, and the immune system. A clinical trial in patients with advanced (non-melanoma) skin cancers treated with anti-PD-1 revealed that non-responders had strong gene signatures for nerve injury and glial cell response to injury in pre-treatment samples. This correlation was unique to "hot", highly immune-infiltrated tumors (no correlation was found in cold tumors), highlighting robust neuro-immune crosstalk. When looking at TCGA data of other anti-PD-1-treated tumors (melanoma and gastric cancer), poor responses to immunotherapy correlated with a strong nerve injury signature. Spatial nanostring genomics revealed signatures of Parkison’s disease, Alzheimer's disease, and ALS in nerves in the tumor microenvironment (TME). Denervation (removal of nerves in the TME) in preclinical models led to better responses to anti-PD-1. In contrast, a procedure called axotomy, in which the nerves were injured, but not removed from the TME, the response to anti-PD-1 was lost. To find out what might have caused nerve injury in the pre-treatment samples of the patients, Amit and his team used scanning electron microscopy and found evidence of perineural invasion, where cancer cells crawl along nerves to metastasize. This is more frequently seen in pancreatic, prostate or salivary gland cancer, and negatively affects prognosis. Cancer cells encasing the axon induce injury by breaking the protective myelin sheath (demyelination), structurally and functionally damaging the nerve, and causing a loss of electrical conduction (signal propagation). In human samples and a perineural invasion model, it was shown that in areas of nerve injury (marked by ATF3 expression, the master regulator of nerve regeneration after injury), the immune microenvironment shifts from functional CD8+ T cells to an accumulation of pro-tumorigenic macrophages and exhausted CD8+ T cells. Non-responders showed elevated type I IFN and IL-6 expression. Immune suppression is a mechanism of anti-PD-1 resistance, and ATF3 drives this resistance by boosting type I IFN. Blocking IFNAR1 restored anti-PD-1 sensitivity and slowed tumor growth in mouse models. Amit finished his talk by summarizing that nerve injury, often caused by perineural invasion, drives an immunosuppressive microenvironment, causing resistance to anti-PD-1. He also pointed out that the nervous system is a highly targetable system, offering new therapeutic avenues by repurposing neurological and psychiatric tools to enhance cancer treatment.
From FMT to Second Generation Microbiome Interventions to Increase Cancer Immunotherapy Efficacy- Bertrand Routy, MD, PhD – University of Montreal
Metagenomic sequencing of fecal samples from immunotherapy patients has consistently shown distinct microbiome compositions between responders and non-responders, leading to the recognition of the gut microbiome as a hallmark of cancer. Bertrand Routy summarized early trials that identified various beneficial bacteria (Akkermansia muciniphila, Ruminococcus, and Bifidobacterium), but his meta-analysis showed that “deleterious bacteria”, enriched in non-responders, were much more consistent across trials. Over 100 studies have now linked antibiotic use to the enrichment of these deleterious bacteria, leading to ASCO guidelines for prudent antibiotic use (and a recommendation against routine use of probiotics). Fecal Microbiota Transplantation (FMT) from non-responder patients to germ-free mice induced resistance to immune checkpoint blockade (ICB), which was reversed by a second FMT from a responder, or from a donor with specific bacteria (e.g., Akkermansia). This established FMT as a new therapeutic avenue, with seven clinical studies showing its safety and potential efficacy in boosting immunotherapy response. Longitudinal analysis of patients who had received FMT plus ICB revealed that responders lost more bacteria over time, primarily deleterious ones like Enterocloster, even though fecal microbiome engraftment rates were similar between responders and non-responders. A proof-of-principle experiment confirmed that adding the lost deleterious bacteria back negated the efficacy of immunotherapy in mice. This bacterial loss correlated with an upregulation of CD69+ CD8+ T cells and decreases in the immunosuppressive tryptophan pathway (loss of kynurenine/quinolinic acid). However, FMT faces many limitations (scalability, donor source and variations, and risk of infections), and novel strategies are needed. Routy then shifted to an alternative strategy centered around the prebiotic polyphenol Castalagin, which transformed PD-1-resistant tumors into sensitive ones by modifying the microbiome and increasing Akkermansia and Ruminococcus counts in mice. Castalagin's effect was found to be dependent on the upregulation of secondary bile acids (DCA, UDCA, muricholic acid), which was accompanied by an upregulation of corresponding liver genes and proteins. The effect was abolished by an FXR antagonist or a bile acid scavenger, and absent in germ-free mice, due to lack of gut bacteria. Castalagin is metabolized into urolithins, with urolithin C showing good anti-tumor activity and also demonstrating secondary bile acid dependency in mice. Further, giving the bile acid ursodeoxycholic acid (UDCA) itself, showed strong anti-tumor activity in multiple mouse models. Based on these findings, Routy and his team initiated a Phase 1 trial combining camu camu (containing castalagin) with ICB in NSCLC and melanoma patients. Among 15 patients with refractory melanoma, they observed one complete response, one partial response, and a 56% one-year overall survival. In NSCLC, progression of the primary tumors was suppressed. Responders showed a significant increase in the surrogate marker, urolithin C.
Reading suggestion:
Trust a first responder’s gut.
Circadian control of the tumor immune microenvironment determines immune checkpoint inhibitor efficacy- Jake Lichterman, DO – UT Southwestern Medical Center
Jake Lichterman presented his work on the role of the circadian clock in regulating the tumor immune microenvironment (TME), and its impact on immune checkpoint inhibitor (ICI) efficacy. The circadian clock is driven by transcription factors like Bmal1 and Clock, and establishes 24-hour rhythms in gene transcription that are critical for immune functions, cell migration, and anti-pathogen responses. In an initial experiment using the subcutaneous MC38 mouse model, anti-PD-1 therapy was administered at six different zeitgeber times (ZT), with ZT0 corresponding to when the lights are turned on, and Z12 corresponding to when the lights are turned off. A significant difference in efficacy was observed with treatment at different ZT. Treatment at ZT2 (early in the light cycle) resulted in the highest efficacy and smallest tumor volumes, while treatment at ZT18 (in the dark cycle) was the least efficacious, with tumor volumes similar to isotype controls and correlating with reduced survival. This time-of-day-dependent effect was recapitulated in three other tumor models, in different tissue locations, and with anti-CTLA-4 treatment. Timing of the first infusion alone was sufficient to dictate the therapeutic outcome. Based on these findings, Lichterman hypothesized that the clock regulates the TME prior to the first ICI dose. Spatial transcriptomics on tumors of identical size revealed more immune cell infiltration at ZT2, with T cells and myeloid cells in closer proximity compared to ZT18. Flow cytometry confirmed a difference in percentage of live immune cells in tumors of identical weight, peaking at ZT2, with the most significant variation in conventional dendritic cells (cDCs) and CD8+ T cells. Mice treated at ZT2 also showed higher numbers of total and activated (IFNγ+ and granzyme B+) CD8+ T cells in the tumor. Conditional Bmal1 knockout mice confirmed the direct involvement of circadian regulators, and knockout of Bma1 specifically in dendritic cells or in CD8+ T cells blunted the anti-PD-1 efficacy observed at ZT2, demonstrating that a functional clock in both cell types is essential for the time-of-day effect. Analysis of FACS-sorted tumor-infiltrating DCs showed upregulation of the chemokine CX3CL1 at ZT2. It was further shown that the clock protein Bmal1 directly binds to the promoter region of the Cx3cl1 gene in DCs. Intratumoral injection of recombinant CX3CL1 at the non-efficacious time (ZT18) rescued the therapeutic effect and increased the infiltration of CX3CR1+ CD8+ T cells. Moreover, the therapeutic benefit of treatment at the optimal time (ZT2) was lost in CX3CR1-receptor knockout mice, validating the importance of the CX3CL1–CX3CR1 axis. Lichterman proposed that the time of day dictates clock-dependent CX3CL1 expression in DCs, which in turn, promotes the infiltration of CX3CR1+ CD8+ T cells, which determines ICI efficacy. Finally, Lichterman presented translational data from renal cell carcinoma patients, which showed that the time of the first ICI infusion (ipilimumab + nivolumab) correlated with clinical outcomes. Patients treated in the morning had almost double the response rate and improved overall survival over five years compared to those treated in the late afternoon.
Reading suggestion:
Non-mutated tumor antigens in the spotlight for therapeutic targeting.
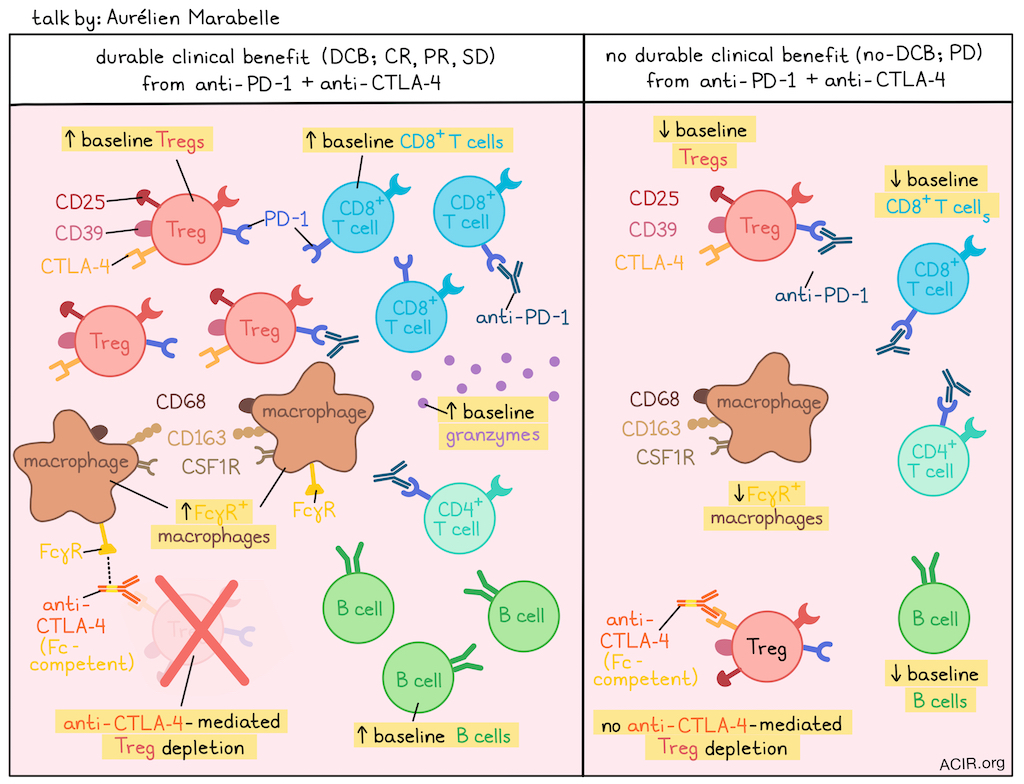
High Tregs and M2 Macrophages in the TME Predict Efficacy of Immune Checkpoint Targeted Immunotherapies- Aurélien Marabelle, MD, PhD – Gustave Roussy
Aurélien Marabelle highlighted that the prognostic value of biomarkers is treatment-dependent, citing the Philadelphia chromosome as a poor prognostic marker for leukemia before imatinib became available, but a key therapeutic target after. He proposed this applies to Tregs and macrophages in the TME, as the long-held belief that Tregs and macrophages are bad was based on studies in patients receiving chemotherapy, not immunotherapy. Marabelle showed that in human tumors, intratumoral Tregs express high levels of CD25 and CD39, and membrane-bound CTLA-4 is selectively expressed on Tregs, whereas PD-1 is broadly expressed on CD8+ T cells, conventional CD4+ T cells and CD4+ Tregs. This underpins their different therapeutic mechanisms: anti-PD-1 antibodies are Fc-silent (IgG4) with no dose, efficacy, and toxicity correlations, whereas anti-CTLA-4 antibodies are Fc-competent (IgG1) with dose, efficacy, and cytotoxicity all being correlated. To maximize the therapeutic index of anti-CTLA-4 therapy, Marabelle introduced a randomized trial in first-line metastatic melanoma comparing standard intravenous (i.v.) anti-CTLA-4 versus a 10-fold lower dose of intratumoral (i.t.) anti-CTLA-4, with all patients receiving standard i.v. anti-PD-1. The i.t. arm was extremely safe, meeting its primary safety endpoint. In terms of efficacy, although the trial was not powered to compare arms, analysis within the i.t. group showed a 65% response rate in injected lesions, and a 50% response rate in non-injected lesions, with the i.t. arm demonstrating more complete responses than non-injected target lesions. Translational analysis of patients pooled from both arms based on durable clinical benefit (DCB; complete response, partial response and stable disease) at 6 months revealed expected and unexpected biomarkers. As expected, the DCB group had higher baseline CD8+ T cells, B cells (which correlated with CD8+ T cells), and granzyme signatures. The unexpected finding was that the DCB group also had significantly higher baseline levels of intratumoral activated Tregs. Upon treatment, the Treg population was effectively depleted in the DCB group, leading to an increased CD8/Treg ratio. However, Tregs were not depleted in the no-DCB group, supporting the notion that Fc-competent anti-CTLA-4 works in part via Treg depletion. Investigating the mechanism of depletion, Marabelle showed it was not dependent on the level of CTLA-4 expression, which was similar, or even inched higher in the treated no-DCB group. The major difference was a significantly higher expression of Fc-gamma receptors (FcγR), such as CD16 and CD64, in the DCB group. These FcγRs were not found on NK cells, but on macrophages expressing higher levels of CD68, CD163, and CSF1R in the DCB group. This finding was validated in other cancers. A prospective study on resected hepatocarcinoma found that cytotoxic tumors (predicted to respond to checkpoint blockade) also had significantly increased signatures of both Tregs (CD25, CTLA-4) and macrophages expressing high levels of FcγRs (CD16, CD64), CD163, and CSF1R. A separate single-cell RNAseq dataset from non-small cell lung cancer linked the cells in macrophage clusters expressing CD163 and CSF1R with FcγR expression. Marabelle concluded that a high baseline presence of both Tregs and FcγR-high macrophages are indicative of tumors where rejection and tolerance are both operative and thus good predictive biomarkers of response to anti-PD-1 and anti-CTLA-4 therapy, where anti-CTLA-4 therapy leverages FcγR-high macrophages to deplete the CTLA-4-positive Tregs and tip the balance toward antitumor immunity.
Reading suggestion:
Rotavirus vaccine and anti-CTLA-4: a dynamic duo.
T cell and NK cell therapies
Cell Therapy 2.0: Integrating Metabolic Engineering for CAR-T Cells- Julian Lum, PhD – BC Cancer Research Centre
Understanding metabolic barriers to effective cell therapy could lead to new ways to overcome such barriers, and so Julian Lum asked a simple question: are immune cells in tumors metabolically bankrupt (i.e., lacking key metabolites) or do they undergo metabolic arrest due to their metabolic milieu, or both? To address this question, ascites samples from patients with ovarian cancer were used as source material for immune and tumor cells. The tumor interstitial fluid was also obtained and analyzed in detail using mass spectrometry, and used for in vitro assays. Gene engineering was used to test hypotheses. To follow the fate of the key nutrient glucose, 13C-glucose was given as a bolus to patients, and the ascites fluid collected over time. 13C-glucose and 13C-glucose-derived metabolites reached a steady state quickly (within 2 hours). Culturing T cells in ascites fluid showed a depression in IFNγ production, despite high and deemed sufficient levels of extracellular glucose. Looking in the cells, the enrichment of intracellular 13C-glucose was lower in CD8+ T cells, but equivalent levels were observed in tumor and CD4+ T cells. Glycolysis up to pyruvate is similar in T cells and tumor cells; however, the immune cells showed less flux of pyruvate into the TCA cycle (a lower citrate/pyruvate ratio). Thus, nutrient availability did not correspond to uptake, and uptake did not directly correlate with assimilation, but varied depending on the cell type. A lower level of measured isocitrate dehydrogenase (IDH) activity in CD4+ T cells, slowing the conversion of isocitrate to α-ketoglutarate, helped to explain the lower flux to citrate, but citrate actually accumulated in the T cells, so what was the source? Lum’s in vitro system allowed analysis of metabolite uptake from the ascites fluid and showed uptake of lactate, citrate, and malate. Further, citrate specifically suppressed IFNγ production. Citrate is a known allosteric inhibitor of phosphofructose kinase (PFK) to slow down glycolysis. Overexpression of IDH2, which reduces cellular citrate, restored PFK activity and normal glycolysis. In support of these results, expression of a PFK mutant that is insensitive to citrate inhibition demonstrated normal IFNγ production in the presence of high citrate levels. Expression of IDH2 in CAR T cells enhanced their IFNγ production and cytolytic activity in vitro, suggesting an opportunity for effective metabolic engineering of T cells.
Reading suggestion:
Metabolic characteristics of DC vaccines define therapy responses.
CRISPR screens reveal innate immune checkpoints regulating NK cell sensitivity across solid tumors- Haolong Li, PhD – Fred Hutchinson Cancer Center
Prostate cancer (PCa), the most common cancer among males in the U.S., shows poor response to immune checkpoint blockade due to its "immune-cold" nature, including dysfunctional T cells, M2 macrophages, and low MHC-I expression. Haolong Li explored NK cells as an MHC-I independent mechanism for killing PCa cells. However, NK-92 cells failed against PCa lines (but not other tumor cell lines), suggesting a protective mechanism by the tumor cells. To identify vulnerabilities, Li and his team used an unbiased, high-throughput CRISPR screen. Initially, a genome-scale CRISPR screen did not yield results, even for expected targets, and the screen was refined by using a pan-cancer surface receptor (‘surfaceome’) CRISPR library with curated controls across six cancer types (12 cell lines). The screen confirmed canonical inhibitory hits, especially genes in the antigen presentation pathway, (e.g., NECTIN2, PTPN2, TAP1/TAPBP/HLA-B) and established activation pathway hits (STAT/IFNγ signaling). The most significant novel hit was the solute carrier SLC7A5, which is highly expressed in tumors and negatively correlates with NK activity. In vitro validation showed that SLC7A5 knockout in tumor cells enhanced NK activation and degranulation. In vivo, NK-92 cells were able to dramatically slow the growth of SLC7A5 KO tumors compared to wild-type SKOV3 tumors, with a large fraction of the luciferase marker disappearing just one day after NK cell infusion. Of note, a clinical-grade inhibitor of SLC7A5 already exists.
Beyond
SARS-CoV-2 mRNA vaccines sensitize immunologically “cold” tumors to immune checkpoint blockade- Adam Grippin, MD, PhD – The University of Texas MD Anderson Cancer Center
Based on his prior work showing that RNA/LNP vaccines coding for non-tumor antigens could enhance the efficacy of immune checkpoint blockade (ICB) in cancer treatment, Adam Grippin hypothesized that COVID-19 mRNA vaccines may be similarly effective. Retrospective studies in NSCLC, metastatic melanoma, and a broad cancer cohort at MD Anderson showed significantly improved survival (overall and/or progression-free) for patients who received a COVID-19 mRNA vaccine, within 100 days (most pronounced within 30 days) of starting ICB. This benefit was not seen with other non-mRNA vaccines (influenza or pneumonia) with ICB or for COVID-19 mRNA vaccines with chemotherapy. In models of established B16F0 or Lewis lung cancer, the vaccine plus ICB inhibited tumor growth and induced a robust surge of IFNα, which was not observed with the packaging lipid nanoparticle particle (LNP) alone. Further, the benefit of the COVID-19 mRNA vaccine was eliminated with blockage of the IFNα receptor. Grippin and his team could show that IFNα activated myeloid cells (DCs and macrophages) in lymphoid organs and tumors, and that these activated cells primed functional T cells with specificity for the tumor models and for patients’ relevant tumor-associated antigens. These T cells infiltrated the tumor, and Grippin was able to demonstrate a correlation of their activity and compensatory PD-L1 upregulation on tumor cells, making the tumor more responsive to ICB. In healthy human volunteers, the COVID-19 mRNA vaccine stimulated a similar innate immune response activation, and a significant upregulation of IFNα, leading to the increased activation of CD11b+ myeloid cells, CD11c+ DCs, CD56+ NK cells, and CD8+ T cells. As a surrogate marker for the activation of effective T cells, Grippin looked at PD-L1 expression on tumor cells in patients with NSCLC who had received ICB and the COVID-19 mRNA vaccine. He and his colleagues found an increased PD-L1 tumor proportion score (TPS) in patients who had received the vaccine within 100 days prior to biopsy compared to in patients who had not received the vaccine or received vaccine more than 100 days prior. The vaccine's advantage in survival was observed even in immunologically “cold" lung cancers (PD-L1 TPS < 1%), suggesting a strategy to sensitize cold tumors to ICB. A randomized clinical trial is planned to definitively validate this effect, and efforts are underway to develop next-generation universal immune stimulants to prepare the immune response to ICB.
Reading suggestion:
COVID-19 mRNA vaccines provide a surprising boost to ICB therapy efficacy.
More Than Cell Killers: Mechanistic Insights into ADC Payload Classes and Immune Engagement- Margaret K. Callahan, MD, PhD – University of Connecticut School of Medicine
Margaret Callahan gave a broad overview of the myriad alternatives that have been and are now being developed to optimally engineer antibody drug conjugates (ADC) as a therapeutic approach. The three key structural components of the ADC include the antibody or antibody-like domain that confers specificity, the cytotoxic payload, and the structure and chemistry of the linker between the antibody and the payload. The functional impacts that Callahan focused on following ADC delivery were localization of the ADC (and payload delivery) to the tumor, including fine targeting to immune, tumor, or both cell types, and the impact of the payload on the immune system, including the induction of immunogenic cell death (ICD). The danger theory of immune response posits that antigen alone is insufficient to activate immunity, but that a variety of signals released from stressed, dying or dead cells (commonly ATP, surface calreticulin, and type I IFN and IL-1β cytokines) complement the antigen signal and generate a T cell response. ADCs seem capable of initiating ICD and stimulating antitumor immunity, and particular payloads, such as vedotin and deruxtecan, are more potent, although more rigorous testing is needed. ADCs have grown beyond only tumor markers, and important new cell targets to potentially eliminate are immune cells. These targets include CD25 (marking Tregs), B7-H3, and PD-L1 (marking inhibitory myeloid cells) and cells expressing the nucleotidase CD73, which contributes to immunosuppressive adenosine. Immune activation can be helpful, but can also contribute to systemic toxicities, and some efforts in the clinic have been halted due to toxicity. Thus, significant efforts are underway to optimize the balance. Consequently, immune-stimulating antibody conjugates (ISACs) carrying payloads, such as immune stimulating TLR- or STING-agonists have been developed. A recent ISAC, tested clinically, targeted a TLR7/8 agonist to HER-2 expressing cells. It showed some activity and a relationship between immune activation and outcome, but was halted with hopes of finding a molecule with a better therapeutic index. Taking this concept a step further, one trial targeted CD22 on B cells with a proprietary CpG analog as the payload, combining immune cell targeting and stimulation. Some durable responses and limited toxicity were encouraging readouts. Callahan closed by pointing to the intrinsic and, through engineering, added immune-activating properties of ADCs, how this can be optimized to deliver the necessary functional effects, such as ICD, and how these effects can be modulated by the tumor cells or the TME. A wide range of tools, approaches, and new understanding can now be applied to optimize clinical results with ADCs.
Reading suggestion:
ESMO Targeted Anticancer Therapies Congress 2024.
Ivonescimab: A Novel First-in-Class PD-1 x VEGF Bispecific Antibody- Allen S. Yang, MD, PhD – Summit Therapeutics
Bispecifics are heterozygous antibodies that bind two specific targets and can be used as T cell engagers, dual checkpoint inhibitors in the same cell, or dual agents targeting different cells. Allen Yang focused on ivonescimab, a novel bispecific antibody that consists of an Fc-null bevacizumab (anti-VEGF) backbone fused at its C-terminus to the scFvs from penpulimab (anti-PD-1). Yang highlighted that the molecule’s cooperative binding enhances its affinity and makes it more than the sum of its parts. This cooperativity was demonstrated in vitro: the binding of ivonescimab to PD-1 was increased 18-fold in the presence of VEGF, and conversely, its binding to VEGF was increased 4-fold in the presence of PD-1. These changes are driven mostly by the off-rate, meaning the presence of both ligands locks the molecule in place. The antibody is therefore hypothesized to be enriched in the TME, where both PD-1 and VEGF are co-localized at high concentrations, potentially increasing efficacy. A second key feature of ivonescimab is the tetravalent format. The two binding domains for the VEGF form a cross-linked structure via the dimeric VEGF molecules, daisy-chaining ivonescimab and increasing the valency for targeting PD-1 on lymphocytes. This enhances activation of lymphocytes, potentially increasing their antitumor efficacy while reducing on-target, off-organ toxicity. Pharmacokinetically, ivonescimab has a shorter half-life (9-10 days) than bevacizumab (~3 weeks). Though dosed at a molar equivalent to bevacizumab (20 mg/kg), and achieving similar peak concentrations, this results in lower trough concentrations, providing a respite from VEGF suppression that may further improve its safety profile. Globally, 14 phase 3 trials for ivonescimab are underway by Summit therapeutics or Akesobio. Yang noted the drug's promising activity in non-traditional PD-1 or VEGF target indications, such as microsatellite-stable colorectal cancer and pancreatic cancer. Yang focused on the HARMONY-2, a double-blind, phase 3 study comparing ivonescimab monotherapy to pembrolizumab monotherapy in locally advanced or metastatic treatment-naive NSCLC. ivonescimab almost doubled the median PFS compared to pembrolizumab (11.1 vs. 5.8 months; HR 0.51) and showed equivalent efficacy across both squamous and non-squamous histologies. Importantly, ivonescimab was superior to pembrolizumab not only in the PD-L1-low (PD-L1 TPS 1-49%) population, but also in the PD-L1-high (PD-L1 TPS >50%) and even very high (PD-L1 TPS >90%) group. Yang argued these results clinically validated the hypothesis that the ivonescimab 's unique cooperativity provides a benefit beyond simple co-blockade. While Grade 3 adverse events (AEs) were higher for ivonescimab, serious AEs such as AEs leading to discontinuation or deaths were equivalent to pembrolizumab. VEGF-related AEs (proteinuria, hypertension) were higher, as expected, but manageable. Yang concluded that ivonescimab is a first-in-class PD-1 x VEGF bispecific developed for a broad range of cancers with increased cooperativity driving its efficacy and that the data from other agents with different bispecific formats will shed more light on the mechanisms of these molecules.
Reading suggestion:
Masking bispecific T cell engagers until they reach the tumor improves their toxicity profile.
By Ute Burkhardt, Ed Fritsch, Shishir Pant, and Lauren Hitching.



