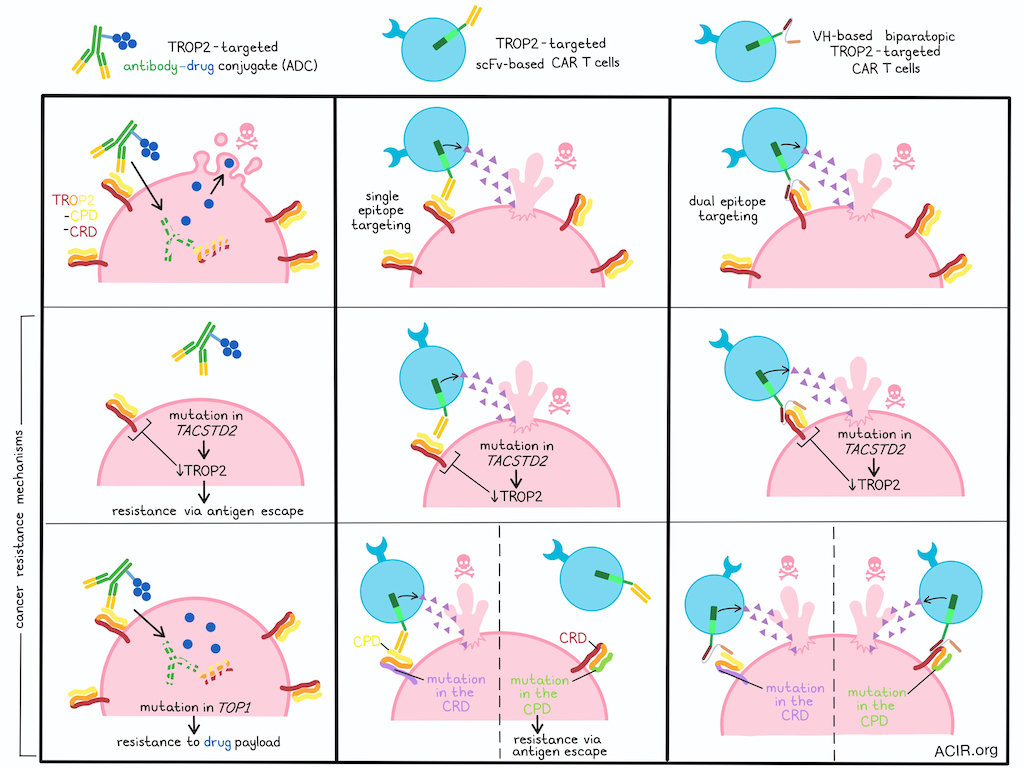
TROP2 is a fetal oncogene that is expressed in a number of cancer types, and while it can be successfully targeted by antibody–drug conjugates (ADCs), cancer cells often develop ADC resistance, either through loss of expression of the targeted TROP2 epitope, or acquired resistance to the drug payload. In an effort to more successfully target TROP2, Brea et al. engineered TROP2-targeted CAR T cells, including biparatopic CAR T cells targeting multiple TROP2 epitopes to minimize antigen escape. Their work was recently published in Cancer Immunology Research.
To begin, Brea et al. designed second-generation CARs utilizing scFvs derived from the antibody sacituzumab-govitecan (clone hRS7, approved under the brand name Trodelvy), along with 4-1BB costimulatory and CD3ζ signaling domains. In vitro, CAR T cells expressing these CARs successfully targeted TROP2+ cells, including the EGFR-mutant NSCLC cell lines PC9 and HCC827GR6, and the TNBC cell lines HCC70 and MDA-MB-231. CAR T cell cytotoxicity was also maintained against HCC827GR6 cells that had acquired resistance to the EGFR inhibitor osimertinib, and in 3D culture models of PC9.
In NSG mice bearing established PC9 or HCC827GR6 tumors, a single dose of TROP2-targeted CAR T cells induced regression, even at low doses. Responses were durable until 75 days, with some mice succumbing to what appeared to be graft-versus-host disease rather than tumor burden. Similar efficacy was observed in a model of metastatic lung tropic PC9, in which low doses of CAR T cells induced antitumor activity in 80% of mice.
Moving to more clinically relevant models, the researchers evaluated their TROP2 CAR T cells against tumor spheroids of an established patient-derived EGFR-mutant NSCLC sample, as well as against organotypic spheroids of PC9 and HCC827GR6, and observed substantial in vitro activity, including increased IFNγ levels correlating with cytotoxicity. Similarly, in two patient-derived xenograft models of EGFR-mutant NSCLC, a single dose of CAR T cells induced significant tumor regression. These effects were dependent on TROP2 expression.
In previous studies, TROP2-expressing cancers have developed resistance to TROP2-targeted ADCs through mutations in TACSTD2 (encoding TROP2) that decrease TROP2 expression, and mutations in TOP1 (encoding topoisomerase) that support resistance to ADC payloads. To further study these mechanisms of resistance and the potential to overcome them, Brea et al. developed PC9 cells expressing WT TROP2, mutant T256R TROP2 (reducing TROP2 expression), or mutant E418K TOP1. Expression of either mutant gene led to resistance to TROP2 ADCs, but these cells remained sensitive to TROP2 CAR T cell-mediated activity, even when TROP2 expression was downregulated by the T256R mutation.
While TROP2-targeted CAR T cells derived from scFVs appeared to outperform ADCs, Brea et al. sought to further improve their efficacy by developing TROP2-targeted CARs from heavy chain (VH)-only domains, which are smaller than scFvs and can more easily be linked in order to target multiple epitopes. To do this, a number of VH-only binders were derived from heavy chain-only human antibody (HCAb) mice, incorporated into CAR constructs, and tested for activity against PC9 and HCC827GR6 cells. Constructs that induced high levels of cytotoxicity were then further studied in a lung metastasis model, where many of the VH-only CAR T cells performed as well as the scFv-derived hRS7 CAR T cells. The VH681-based CAR emerged as a lead candidate, demonstrating the strongest tumor control and improved survival in mice.
In prior research, sacituzumab and the scFvs derived from it have been found to bind to residues Q237-252 in the cysteine-poor domain (CPD) of TROP2, as did the antibody datopotamab deruxtecan (approved under the drug name Datroway). Screening the binding of a variety of VH-only binders, the researchers substituted the domains of human TROP2 with their murine counterparts (which demonstrate low cross-reactivity to the human versions) to determine VH binding, and found that VH681 bound a unique region of TROP2 in the cysteine-rich domain (CRD), thus distinct from the CPD binding of approved ADCs. Computational modeling of protein–protein interactions further identified key putative residues on TROP2 that formed the epitopes for each VH binder.
Next the researchers took advantage of the fact that VH-only binders are easy to link for dual targeting, and developed biparatopic CARs that simultaneously targeted two unique epitopes of TROP2 – one in the CPD (VH375) and one in the CRD (VH681). In cytotoxicity assays against PC9 expressing TROP2 substituted with either a murine CPD or CRD domain, only biparatopic VH binder CAR T cells (versus CAR T cells utilizing the hRS7 scFv or single-VH constructs) maintained cytotoxicity in both settings, expressing elevated levels of pro-inflammatory cytokines and cytotoxic effectors, including IFNγ, IL-2, TNFα, perforin, and granzyme B against the TROP2+ target cells. Similar results were observed in a live-cell imaging cytotoxicity assay of various TROP2-targeting constructs against PC9 cells with various murine substitutions in TROP2 domains. Computational modeling further validated that the biparatopic CARs effectively targeted both the CPD and CRD domains simultaneously.
Finally, the researchers evaluated their biparatopic CAR T cells in NSG mice engrafted on one flank with PC9 with the murine CPD substituted, and on the other flank with PC9 tumors with the murine CRD substituted. While CAR T cells containing the CPD-targeted hRS7 scFv effectively cleared PC9 tumors only on the flank with the CRD domain substituted, the biparatopic VH681_375 CAR T cells mediated durable and effective tumor control on both flanks and extended mouse survival. These results suggest effective co-targeting of the CPD and CRD of TROP2 by the biparatopic CARs. Further, no changes in weight were observed, suggesting limited toxicity.
Overall, these results suggest that CAR T cells can be used to target TROP2, including in settings that are resistant to ADCs through either antigen loss or resistance to the drug payload. Further, engineering biparatopic CAR T cells with VH-only domains, rather than typical scFvs, allowed for successful co-targeting of two unique epitopes on different domains of TROP2, which could further limit antigen escape that occurs through mutation or loss of target epitopes, and could potentially help to prevent resistance in a clinical setting in the future.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Elliott Brea answered our questions.

What was the most surprising finding of this study for you?
We became interested in targeting cell-surface molecules like TROP2 in solid tumors, given the highly prevalent expression on multiple solid tumors, such as lung cancer, breast cancer, and bladder cancer, as well as track record for TROP2 being targeted by antibody–drug conjugates (ADCs). We were surprised by how effective the CAR T were against TROP2+ solid tumors, including in models where ADC failed. We think this opens the possibility for TROP2-targeted CAR T cells as a strategy for eliminating TROP2+ solid tumors, including after progression on TROP2-targeted ADCs.
What is the outlook?
Encouraged by these findings, we successfully secured a Team Science Award from the Lung Cancer Research Foundation and the International Association for the Study of Lung Cancer to build on this work. With this support, we’re now designing the next generation of CAR T cells that can more precisely target tumor cells while avoiding healthy tissues. One strategy we're particularly excited about is “logic-gated” CAR T cell design — a sophisticated approach that allows the cells to become active only in the presence of a unique combination of tumor-specific markers.
This research is still in its preclinical stages, but we hope if successful to develop a safe, effective, and long-lasting therapy that could be combined with existing treatments to prevent relapse, and offer a potential cure for patients with EGFR-mutant lung cancer.
What was the coolest thing you’ve learned (about) recently outside of work?
We have been teaching my 2 year old daughter about all kinds of animals, and we recently visited Bermuda. It was amazing to see her reaction to seeing a real live turtle that weighed almost 400 pounds! It felt like how we read and learn about different techniques and methods in science, and then when we apply it and see it work, the connections we make and then build on are a key part of why science is so fun.



