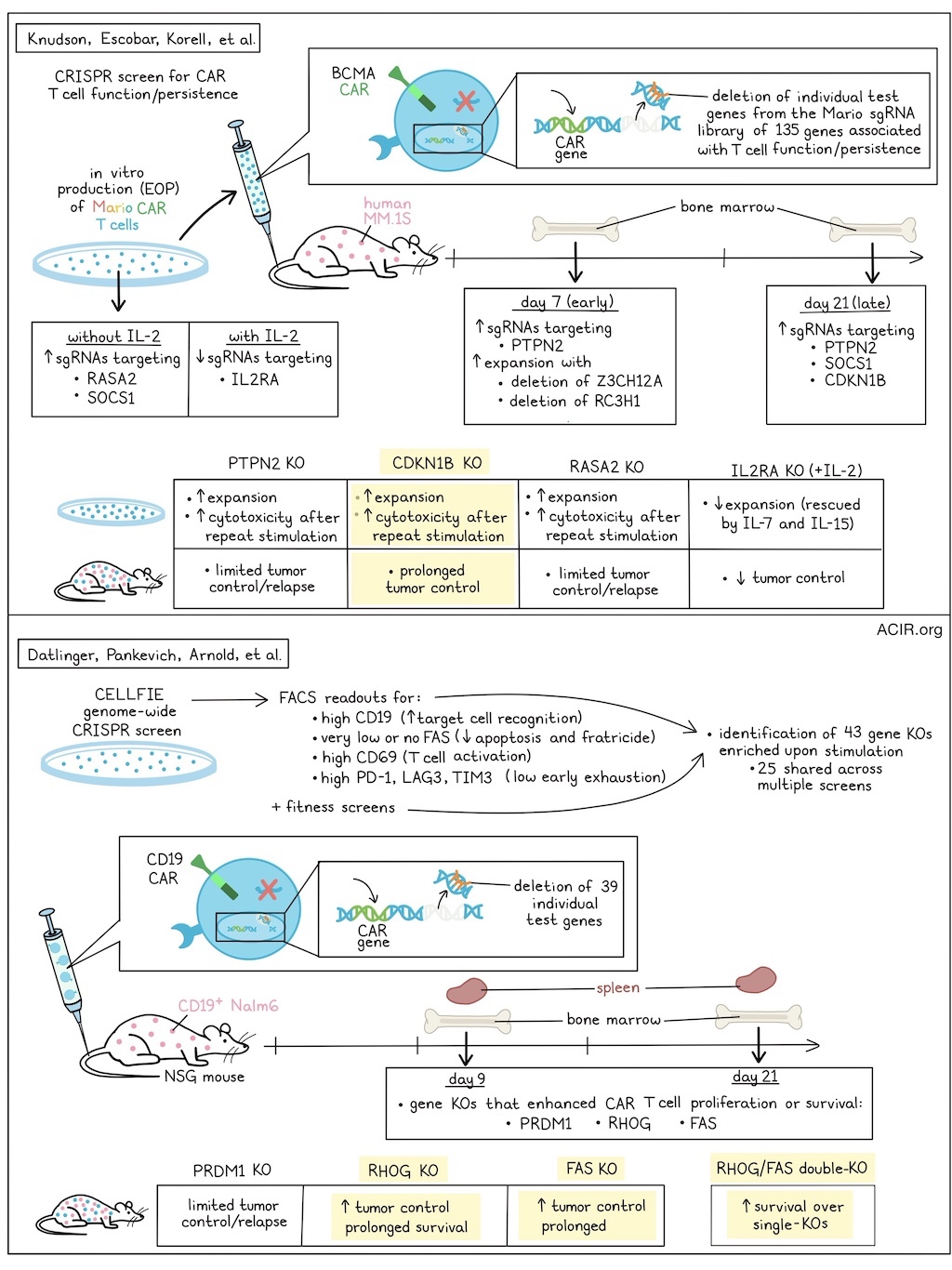
One of the major questions in CAR T cell therapy research is how to improve CAR-T expansion, persistence, and functionality to improve clinical responses and prevent relapses. Two recent back-to-back publications in Nature used CRISPR screening methods to detect genetic targets for improving CAR-T therapy. In the first paper, Knudson, Escobar, Korrell, et al. performed in vivo loss-of-function CRISPR screens in CAR-T to determine which genes contribute to CAR-T persistence and function. In the second paper, Datlinger, Pankevich, Arnold, et al. used a genome-wide in vitro CRISPR screening platform (CELLFIE) to determine genes related to CAR-T function.
Knudsen, Escobar, Korell, et al. used their screening platform to detect genes impacting expansion and early and late persistence of human BCMA-targeting CAR-T in a murine model of human myeloma (MM.1S). A CRISPR single-guide RNA (sgRNA) library (named Mario) was developed, which included 135 genes with known or proposed T cell functions. The sgRNA was delivered to T cells with a separate vector from the CAR, to allow for evaluation of CAR- or target-specific dependencies. To evaluate the effects of genetic changes in the CAR-T on in vivo expansion and persistence, cells were isolated from bone marrow at 7 (early) or 21 (late) days after CAR-T injection and compared to cells at the end of in vitro production (EOP). EOP Mario CAR-T were enriched in sgRNAs targeting RASA2 and SOCS1, and in CAR-T expanded in IL-2, a depletion of sgRNAs targeting IL2RA was detected.
To determine which genetic modifications improve T cell persistence in vivo, the abundance of sgRNAs at early or late in vivo timepoints was compared to the EOP Mario CAR-T. Early in vivo, the most highly enriched sgRNAs targeted PTPN2. Further, deletion of ZC3H12A and RC3H1 also resulted in improved expansion. At the late time point, sgRNAs targeting SOCS1 or PTPN2 were enriched. Despite strong enrichment at EOP, sgRNAs targeting RASA2 had no benefit to in vivo expansion. Additionally, sgRNAs targeting CDKN1B had limited enrichment at EOP, but had the largest enrichment at the late in vivo stage. Similar results were obtained between various manufacturing approaches.
Next, the researchers assessed how enriched and depleted gene deletions impact the T cell transcriptional profile and their frequency after 21 days in vivo using Perturb-seq, which combines CRISPR-based genetic perturbations with scRNAseq. After 21 days, modified CAR-T were isolated from the tumor site (bone marrow) of treated mice for analysis. Focusing on the cells with a single genetic perturbation per cell showed that the transcriptional profile of the CAR T cells varied for each gene deletion, although all were selected based on improved in vivo CAR-T persistence.
To determine which genetic changes generated CAR-T with favorable in vitro expansion and in vivo persistence, the researchers focused on four genes: PTPN2, CDKN1B, RASA2, and IL2RA. RASA2-, PTPN2-, and CDKN1B-KO CAR-T expanded more than controls in vitro, while IL2RA-KO CAR-T cultured with IL-2 expanded less, though this expansion could be rescued by stimulation with IL-7 and IL-15. There were no significant differences in in vitro cytotoxicity between the CAR-T. In an in vitro chronic antigen exposure model, CDKN1B-, PTPN2-, or RASA2-KO CAR-T had preserved cytotoxicity over multiple rounds of stimulation, suggesting that the loss of these genes enhanced the cytotoxicity and prevented dysfunction during repeated antigen exposure.
Next, the various KO CAR-T cells were assessed in tumor-bearing mice. CDKN1B-KO CAR-T induced prolonged tumor control, PTPN2-KO CAR-T induced limited control and resulted in relapses.RASA2-KO CAR-T also resulted in relapses, similar to controls, while the IL2RA-KO CAR-T lacked tumor control. The antitumor responses and improved survival induced by CDKN1B-KO CAR-T were confirmed in another myeloma model, and CDKN1B-KO CAR-T derived from patients with multiple myeloma had similar antitumor functionality in two xenograft models, all pointing to CDKN1B ablation as a potentially valuable approach for future CAR design.
In the second study, Datlinger, Pankevich, Arnold, et al. developed the platform CELLFIE, comprising the Brunello genome-wide guide library, a single lentivirus-delivered vector containing both a CAR (19-BBz CAR) and the guide library (CROP-seq-CAR), and mRNA to deliver a CRISPR/Cas9 editor in primary human T cells. Following the editing period, cells were stimulated either through the CAR or the endogenous TCRs (anti-CD3/CD28 beads), resulting in a pool of T cells with high CAR expression and a sequencing-based readout of gRNA-encoded perturbations. Screens were performed on CAR-T derived from four donors. Using the algorithm MAGeCK, both known and potentially new boosters of CAR-T therapies were identified based on strong enrichment upon stimulation.
The researchers used FACS-based screening readouts for four biological processes that limit CAR-T therapy. This included populations that expressed 1) high CD19 acquisition by target cell trogocytosis (increased target cell recognition), 2) high CD69 (strong T cell activation), 3) very low or absent FAS (reduced apoptosis and fratricide), and 4) PD-1, LAG3, and TIM3 (low early exhaustion). When these screens were analyzed together with fitness screens (monitoring enrichment or loss of guide RNAs following CAR or TCR stimulation), the researchers were able to identify 43 gene knockouts from 58 genome-wide screens, of which 25 were shared across several screens.
To validate the identified hits, CELLFIE was extended to support pooled screens in mice with xenografts. NSG mice were injected with CD19+ NALM6 cells, and on day 5, CD19-targeting CAR-T containing a pooled library of 39 perturbations (8 guide RNAs for each perturbation with unique molecular identifiers to increase the statistical power) were injected. On days 9 and 21, spleen and bone marrow were collected, and the gRNAs were sequenced to determine which gene knockouts enhanced CAR-T survival and proliferation. These screens identified FAS, PRDM1, and RHOG gene knockouts as boosters of CAR-T functionality. While FAS and PRDM1 have previously been shown to impact CAR-T efficacy, RHOG, which encodes a GTPase, was a new finding. Paradoxically, it has been linked to immunodeficiency, and RHOG gene loss is detrimental for normal immune function. However, the RHOG KO in CAR-T had a strong beneficial effect; the cells rapidly expanded and accounted for 25% of all CAR-T 21 days after infusion. Using the CELLFIE process also allows for the delivery of CRISPR base-editing editors instead of CRISPR gene-deleting editors (such as CRISPR/Cas9). With base editors, the researchers identified key functional domains in RHOG.
Evaluating the target genes identified in their screens, the researchers treated leukemic mice with single-gene-KO CAR T cells. While PRDM1-KO CAR-T induced better tumor clearance than WT CAR-T, it could not prevent relapse or death. RHOG-KO and FAS-KO CAR-T, on the other hand, induced tumor clearance and resulted in sustained responses, improving survival. RHOG-KO CAR-T, isolated from spleen and bone marrow on day 15 after injection in a murine NALM6 xenograft model, expressed lower levels of LAG3, TIM3, and TIGIT than WT CAR-T. The researchers then engineered RHOG and FAS double-KO CAR-T, which resulted in better survival than either single-KO. These results were validated in patient-derived CAR-T and across multiple CAR designs.
Overall, the data in these two studies reveal that these novel CRISPR screening methods can help find gene editing targets in CAR T cells to improve their in vitro expansion and in vivo persistence and functionality. The targets derived in these studies, and potential future findings in other model systems, could be used as new study directions to improve CAR-T responses in patients with various cancer types.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first author Nelson H. Knudsen on “In vivo CRISPR screens identify modifiers of CAR T cell function in myeloma” and co-first author Paul Datlinger on “Systematic discovery of CRISPR-boosted CAR T cell immunotherapies” answered our questions.

What was the most surprising finding of this study for you?
NHK: Initially, it was technically challenging for us to perform robust genetic screens in T cells in vivo, and it took some trial and error to optimize the correct experimental setup. Once we had generated some data, we were surprised to find that gene knockouts that enhance CAR T cell persistence in vivo do not necessarily make them more proliferative in a cell culture dish, and vice versa. There are likely a multitude of environmental factors influencing T cell behavior in vivo, such as nutrient availability, growth factor and cytokine signaling, and antigen density. For this reason, it will be extremely valuable to continue discovery efforts for CAR T cell enhancements using in vivo models of human disease to better predict therapeutic success.
PD: The first was that RHOG knockout, which causes a human immunodeficiency, could be beneficial in CAR T therapy. This was a very unexpected finding that highlights the value of unbiased screens like ours. The second was how strong synergistic combinations of knockouts (like our RHOG-FAS combination knockout) could be. This makes us optimistic that we can make a huge leap forward in CAR T therapy by rationally combining the many gene perturbations that individual labs have identified. T cells were never evolutionary optimized for immunotherapy; it's really a hack and not their intended natural function. So in a way, systematic screening, like in our project, can serve as artificial evolution for T cells. The third surprising finding was how effective most CRISPR editors were in T cells, e.g., the base editors and even CRISPRa. Genetic screening in T cells is really only starting. Finally, the fourth surprising finding was that the biggest challenge was to develop an effective in vivo screen that uses UMIs for clonal tracking and can detect gRNAs effectively (through in vivo CROP-seq that uses mRNA instead of DNA). but this was absolutely essential, since large in vitro cultures are far from the reality of CAR T therapy.
What is the outlook?
NHK: The immediate outlook for ourselves and the field is to expand in vivo screening efforts to additional cancers, especially solid tumors where CAR T cell therapy has been less successful. These initial screens characterized persistence and expansion, but new experimental designs could identify genetic modifications that limit T cell exhaustion or enhance cytokine production. We are also intrigued by the potential of treating patients with intentionally heterogeneous pools of genetically modified CAR T cells, targeting multiple genes as a way to rapidly prioritize the most effective enhancements.
PD: For the RHOG-FAS knockout combination, we're preparing a first-in-human clinical trial, and are currently actively looking for funding to enable this for the CELLFIE platform. We’re also exploring in vivo screens in more interesting models (e.g., with functional immune systems), as well as much larger combinatorial screens that explore all known CAR T boosters that have been identified individually by various labs. On a personal level, I just started a research group at the Arc Institute of Stanford, UCSF and Berkeley, where we are using CELLFIE for our Virtual Cell Initiative, to build a virtual model of the cell, with a focus on T cell immunotherapy. We'd like to be able to predict the perturbations that lead to preferred T cell phenotypes, like more memory and less exhaustion.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
NHK: My advice would be to find a team of scientists with whom you are excited to discuss your ideas and data regularly. I find it very fulfilling to tackle big problems within a team of engaged scientists from diverse backgrounds, asking important questions and sharing knowledge from different fields. Solving important problems and developing new technology usually comes with a healthy dose of failures, but it’s pretty rare that you can’t learn anything from a failed experiment.
What was the coolest thing you’ve learned (about) recently outside of work?
PD: My 2 kids (Klara is 5 and David is 1) are teaching me something new every day, and it's beautiful to re-discover the world through their eyes.



