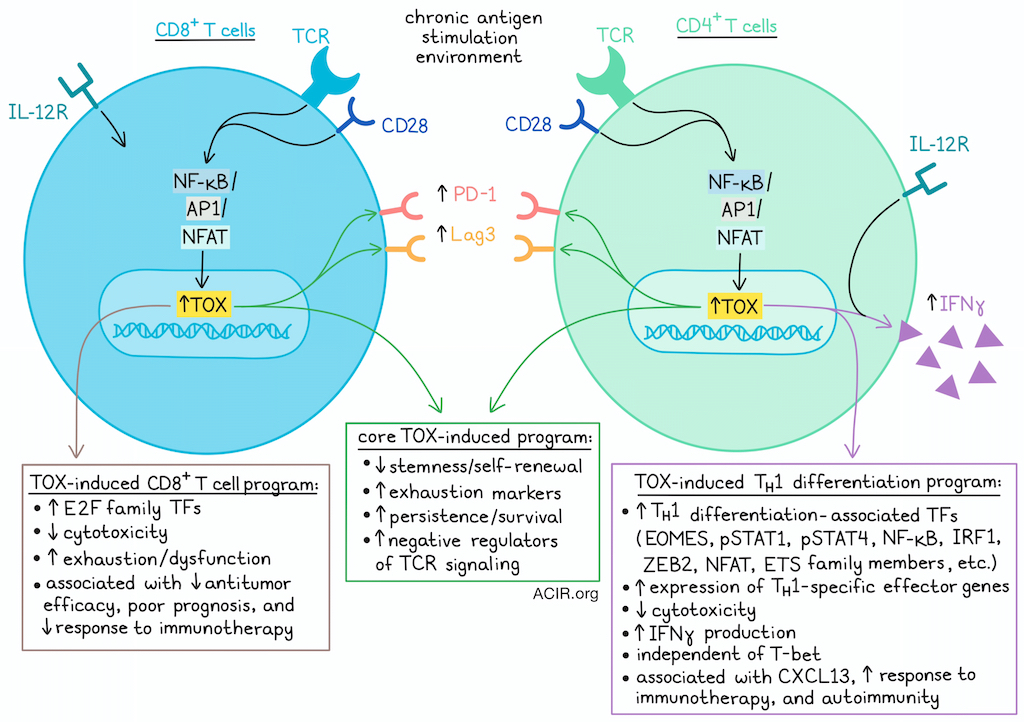
In settings of chronic antigen stimulation, such as chronic infection and cancer, expression of the transcription factor TOX in CD8+ T cells has been associated with cell persistence and survival, but also with exhaustion, dysfunction, and poor prognosis. As the role of TOX in CD4+ T cells is less well understood, Naizir et al. investigated and found that TOX was highly expressed in the type 1 helper (TH1) cell lineage in both mice and humans, and was required for TH1 cell effector function – particularly for the production of IFNγ. Unlike in CD8+ T cells, high TOX expression in CD4+ T cells was associated with improved antitumor immunity and responses to immunotherapy. These results were recently published in Nature Immunology.
To begin, the researchers assessed TOX expression during in vitro polarization of murine naive polyclonal CD4+ splenocytes, activated with anti-CD3 and anti-CD28 and treated with factors to polarize them towards TH1, TH2, TH17, or Treg phenotypes. In both murine and human cells, TOX expression was highest in TH1 cells. In a murine influenza model, antigen-specific TFH cells also showed high TOX expression, and a subset expressed IFNγ.
Evaluating TH1 cells during differentiation, the researchers found that by 48 hours after activation, TOX, T-bet, and EOMES were expressed, and by 72 hours, IFNγ was produced. Both TCR stimulation and CD28 costimulation (signals 1 and 2) were required for TOX induction, and additional IL-12 (signal 3) was required for IFNγ production. Stimulation with either PMA (activating NF-κB and AP1) or ionomycin (activating NFAT) alone was insufficient to induce TOX, but in combination, induced robust TOX expression and expression of T-bet. NFAT was found to be required for TOX upregulation and IFNγ production. Adoptively transferred TCRSMARTA cells (recognizing the LCMV GP61 antigen) in relevant infected or tumor-bearing mouse models also expressed high levels of TOX and produced high levels of IFNγ.
Hypothesizing that TOX may play a functional role in TH1 effector functions, the researchers employed gain- and loss-of-function experiments. Ectopic expression of TOX resulted in a significant increase in IFNγ production, without increasing TNF, IL-17A, or IL-4 (associated with other helper T cell lineages). TCRSMARTA cells from TOX-KO mice had impaired expression of PD-1 and Lag3, and severely impaired production of IFNγ, but not TNF. These cells also showed decreased EOMES, pSTAT1, and pSTAT4, but no changes in markers of other CD4+ T cell phenotypes, suggesting that in chronic stimulation environments, TOX supports TH1 cell polarization and the production of TH1-associated IFNγ in antigen-specific CD4+ T cells.
In tumor-bearing hosts, adoptively transferred TOX-KO TCRSMARTA CD4+ T cells were less abundant by day 21 compared with WT TCRSMARTA T cells, suggesting that TOX supports persistence in CD4+ T cells (as in CD8+ T cells) under chronic stimulation conditions. In TOX-heterozygous (WT/-) TCRSMARTA cells, IFNγ was produced at intermediate levels, and ectopic TOX expression could rescue IFNγ production, suggesting TOX controls TH1 in a dose-dependent manner. Similar results were observed using Cre- and CRISPR-mediated TOX KO models.
Next, the researchers performed RNAseq and ATACseq on WT and TOX-KO TCRSMARTA CD4+ T cells isolated from B16-GP61 tumor-bearing mice, and identified 1,938 differentially expressed genes (DEGs). TOX KO cells showed reduced expression of TH1-specific effector genes (including Ifng and Xcl1) and TH1 differentiation-associated TFs (including Eomes, Nfkb1, Zeb2 and Irf1,, but not TH1-defining Tbth1x21 (encoding T-bet). Gene-set enrichment analysis and gene ontology revealed that while WT cells were enriched for genes and pathways related to TH1 differentiation, TOX KO cells were enriched for genes and pathways related to a stem-like state. ATACseq showed 3,824 differentially accessible regions (DARs) between WT and TOX KO cells, with KO cells showing decreased accessibility and expression of Ifng, Spib, and Nfkb1, and increased accessibility of genes associated with stemness and self-renewal. These results suggested that TOX may promote TH1 differentiation by driving CD4+ T cells to exit their early, lineage-negative state.
While ATACseq did not find DARs within the Tbx21 locus, there were DARs adjacent to the Ifng locus, including reduced accessibility of CNS-34 enhancer (critical for initiating chromatin reorganization of the Ifng locus during TH1 development) and Tmevpg1 (encoding long non-coding RNA Ifngas, a positive regulator of Ifng transcription) in TOX-KO cells, suggesting that TOX may exert epigenetic control over Ifng in a T-bet-independent manner. In line with this, the accessibility of motifs for ETS TF family members (recently identified as T-bet-independent drivers of TH1 differentiation) was also decreased in TOX-KO cells versus WT. When the researchers performed CUT&RUN on TOX-expressing CD4+ TCRSMARTA cells from B16-GP61 tumor-bearing mice, they observed that TOX bound to a variety of sites associated with increased IFNγ production, including Tmevpg1, Stat4, Stat1, Nfatc2, Nfkb1, and Ets1. The role of NF-κB was further confirmed experimentally.
Next, the researchers compared the role of TOX in CD4+ versus CD8+ T cells by analyzing RNAseq data for shared differences between WT and TOX-KO cells for both lineages. Only 139 DEGs were shared between the two lineages, including reduced expression of genes encoding exhaustion markers and increased expression of genes encoding negative regulators of TCR signaling. TF activity analysis showed results in line with this “core” TOX regulatory program, as well as distinct programs in CD4+ versus CD8+ T cells. TOX CUT&RUN experiments in this setting showed TOX binding to shared sites within stemness-associated genes in both CD4+ and CD8+ T cells. In CD4+, but not CD8+ T cells, TOX also bound to Nfatc2 and multiple drivers of NF-κB signaling. These data suggest that in both CD4+ and CD8+ T cells, TOX is induced downstream of TCR signaling, leading to the repression of stem-associated genes, and allowing T cells to exit their naive/stem-like state through the concerted effects of NFAT, NF-κB, and ETS TFs in CD4+ T cells (versus E2F-family TFs in CD8+ T cells), as well as STAT signaling.
Analyzing published scRNAseq data, the researchers found that in TILs from patients with early-stage hepatocellular carcinoma treated with anti-PD-1, CXCL13-expressing CD4+ T cells (previously correlated with clinical responses in the same data) were more frequent in responders than non-responders, as were TOX-expressing CD4+ T cells. TOX expression was also significantly higher in CXCL13+ CD4+ T cells, which correlated with an improved response to ICB. Further, IFNG expression was enriched in the CXCL13+CD4+ cluster, with a correlation between TOX and IFNG expression on a per-cell basis. Similar patterns were observed in data from bladder cancer treated with anti-PD-L1. In a melanoma dataset, TOX was expressed in both T-bet+ IFNγ-producing TH1 cells and cytotoxic EOMES+granzyme B+ CD4+ T cells.
Investigating whether TOX expression might correlate with pathogenic CD4+ T cell responses in the context of inflammation or autoimmunity, the researchers evaluated scRNAseq data from patients with autoimmune vasculitis, and identified a subset of cytotoxic CD4+ T cells that was associated with disease pathogenesis and expressed high levels of TOX and IFNγ. In an autoimmune mouse model type 1 diabetes, the effector TH1 phenotype associated with disease also showed high expression of TOX compared to naive or polyclonal CD4+ T cells, and produced IFNγ upon ex vivo stimulation. Additionally, IFNγ production was impaired in TOX-KO TFH1 cells.
Overall, these results show that while TOX mediated a core epigenetic program in both CD4+ and CD8+ T cells, it also drove distinct programs. TOX expression in CD4+ T cells supported TH1 differentiation and function, and in contrast to CD8+ T cells, was associated with increased cytotoxicity, antitumor immunity, and responses to immunotherapy, as well as with pathogenic responses in autoimmune and inflammatory diseases in mice and humans.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Brianna Naizir answered our questions.

What was the most surprising finding of this study for you?
The most surprising finding of this study was that TOX exerts completely opposing functions in CD4+ and CD8+ T cells. I think that the previous publications on TOX in CD8+ T cells led to this general sentiment in the field that expression of TOX is a bad thing: it marks CD8+ T cell exhaustion phenotypes and is sometimes associated with reduced responsiveness to immune checkpoint blockade in patients with cancer patients. Embarking on this project, we were interested in understanding whether TOX drives phenotypes in CD4+ T cells as well. Surprisingly, we showed that TOX expression in CD4+ T cells is actually associated with improved CD4+ T cell functionality, including responsiveness to immune checkpoint blockade in cancer, as well as pathogenicity in autoimmune diseases. Therefore, I hope this study challenges the field’s perception of TOX and proves that we need to think about this transcription factor with more nuance, considering the context and cell type in which it is expressed.
What is the outlook?
In this study, we show that expression of TOX in CD4+ T cells is associated with the production of IFNγ, improved antitumor immunity, and immunotherapy responses in patients with cancer. This is in contrast to what has been previously shown in CD8+ T cells in human cancer, where TOX has sometimes been linked to poor prognosis and CD8+ T cell exhaustion. Hopefully, this finding highlights the potential use of TOX expression in CD4+ T cells as a prognostic marker for clinical responses in cancer patients in the future.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
If I could go back in time to give my younger self one piece of advice, it would be to embrace the inexplicable data! I would tend to become discouraged or intimidated by data that did not exactly fit my hypothesis, but I now know that sometimes that can lead to the most exciting discoveries!



