
The ACIR team attended the AACR IO meeting held on February 18-21, 2026 in Los Angeles, CA, USA. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
T cells
Philip D. Greenberg
Rafi Ahmed
Fred Ramsdell
Alexander Marson
Robert G. Newman
Owen N. Witte
Checkpoint blockade
Fathia Mami-Chouai
Dario A. Vignali
Megan J. Priestley
Cancer vaccination
Ross Ward
Tzu-Jiun Kuo
Christopher A. Klebanoff
Cell therapies
Jaime Mateus-tique
Rizwan Romee
Neuro-immune interface
Lili Yang
Immune microenvironment
Lieping Chen
Gary P. Nolan
Max Krummel
T cells
Keynote: Taking T cells where natural evolution hasn’t – sustained function in the TME - Philip D. Greenberg, Fred Hutchinson Cancer Center, Seattle, Washington
With the objective of taking T cell biology to phenotypes and functionality where evolution hasn’t, Phil Greenberg began his Keynote Address by describing the fundamental problem of consistent T cell dysfunction shortly after arrival in tumors. Based on murine data indicating that repeated infusions could improve tumor control, a clinical trial with repeated infusions of T cells engineered with a mesothelin (MSLN)-targeting TCR was initiated, with underwhelming clinical results. Analysis of MSLN-specific blood T cells indicated a proliferative and stem-like/intermediate phenotype, while T cells in the tumor were transcriptionally distinct and had an exhausted signature (high TIM3, low IFNγ) that differed from a classical TGFβ-driven exhaustion signature. Analysis of other tumor types indicated this was relatively unique to pancreatic ductal adenocarcinoma, and was replicated in adoptive transfer of engineered T cells in the KPC model. Spatial transcriptomics identified T cells throughout the tumor region and in TLS-like regions. Cells in the tumor regions showed the TGFβ “exhausted” signature, while those in the TLS-like regions were progenitor exhausted and effector cells, suggesting that the combination of tumor antigen and TGFβ exposure drove this unique exhausted phenotype. TGFβR KO led to more rapid exhaustion, and immune checkpoint blockade was ineffective in reversing this effect. Multiomics and computational analysis of functional and dysfunctional cells identified a list of inferred regulators, and a KO screen identified the transcription factor Aiolos as a key regulator. KO of Aiolos rescued the dysfunctional phenotype, but there were significantly fewer surviving cells, due to a role of Aiolos in suppressing apoptosis. This accentuated the conclusion that tumors utilize multiple pathways to orchestrate T cell dysfunction. Two further strategies were undertaken to overcome dysfunction. One strategy, termed Immunomodulatory Fusion Proteins (IFP; aka switch receptors), fused the external domain of inhibitory receptors to the signaling domain of a stimulatory receptor. Two encouraging examples include fusing Fas to 4-1BB to overcome Fas-induced activation-induced cell death, and fusing TGFβR to the IL-2R signaling domain to create a partial dominant negative TGFβ signal inhibitor. In a similar vein, to provide CD4+ T cell help, which importantly helps to prevent T cell exhaustion, class I TCRs can be added to CD4+ T cells, and the functionality of those CD4+ T cells can be enhanced by adding the CD8αβ co-receptor and an IL-2 signalling fusion protein. A 1:1 mixture of TCR-engineered CD8+ and CD4+ T cells significantly improved tumor control in vitro (with a WT-1 TCR) and in vivo (with a MAGE TCR). A clinical trial with a KRAS G12V TCR engineered into CD8+ and CD4+ T cells is underway. Among the first 3 patients, two patients showed tumor shrinkage, and biomarkers (CA9 and CEA) and ctDNA were reduced. Finally, analysis of transcription across multiple datasets of exhausted cells suggested the E3 ligase was a potential target. A CRISPR KO screen of 78 E3 ligase family members identified Klhl6 as a very important member to maintain functionality and to contain healthy mitochondria. Ectopic Klhl6 expression led to better tumor control and reduced exhaustion in murine and human tumor models, adding it to the armamentarium of potential combination strategies.
What is T cell exhaustion? - Rafi Ahmed, Emory University, Atlanta, Georgia
Rafi Ahmed began his talk with a history of T cell exhaustion, highlighting that it is an active, ongoing, and highly regulated response, sustained through the maintenance of precursor exhausted T cells (Tpex; PD-1+TCF1+Tox+). Investigating the origin and maintenance of Tpex cells in the setting of chronic infection, Ahmed and team showed an early (day 5) bifurcation of naive cells towards either terminally exhausted (Ttex) cells or stem-like Tpex cells. At a later stage (day 45 or 50), these stem-like cells persisted, but showed slightly different profiles compared to their early counterparts, with less proliferation and more quiescent signatures, which could be attributed to the TGFβ signaling. Noting that the bifurcation of naive cells into Tpex or Ttex cells occurs before the system “knows” whether an infection will be acute or chronic, Ahmed compared adoptively transferred cells between acute and chronic infection models. Crucially, early fate decisions were identical in both settings. In the chronic setting, Tpex cells generated in the acute setting integrated with endogenous Tpex cells to sustain their phenotype long-term, while in the acute setting, Tpex cells generated in the chronic setting rejoined the central memory T cell pool. These results suggested that early on during infection, the immune system prepares for both acute and chronic responses simultaneously, and then adjusts accordingly.
In another set of experiments, Ahmed and colleagues investigated the immunogenicity of HPV in treatment-naive patients with HPV+ head and neck cancer (HNSCC) who were eligible for surgical resection. In the blood, even treatment-naive patients showed a relatively high frequency of HPV-specific T cells and evidence of strong CD8+ T cell and B cell responses. Dominant TCRs were found to be shared across T cell subsets (stem-like, effector-like, and exhausted), demonstrating expansion of certain clonotypes. Further, while T cells were fairly responsive to the common E6 and E7 HPV-associated antigens from oncogenic mutations, T cell responses to E2 and E5 antigens were often stronger, with evidence of dominant T cell responses to E2. In tumors, patients showed exceptionally high frequencies of antigen-specific cells, with samples showing as high as over 10% of T cells per tumor showing specificity for a single epitope, though frequencies of HPV-specific CD8+ T cells in the blood were lower. Then, in a phase II clinical trial, patients with early-stage HPV+ HNSCC were treated with two doses of atezolizumab (anti-PD-L1) prior to surgery. After the first dose of atezolizumab (anti-PD-L1), the researchers saw evidence of a proliferative burst of newly arising effector cells generated from stem-like cells. Many of these also expressed increased CD28 and other early activation markers compared to naive cells. Early evidence of pathological responses at the time of surgery was promising, though the study is ongoing. Ahmed is currently working to open additional clinical trials investigating the HPV-specific immune response in the progression of HPV infection and the development of cervical cancers, as well as whether responses to E2 and E5 can be found in patients with cervical intraepithelial neoplasia or cervical cancer.
Reading suggestion:
CD8s get by without a little help from their friends
The bomb to a therapy - Fred Ramsdell, Sonoma Biotherapeutics, Seattle, Washington
Fred Ramsdell, Nobel Laureate in Physiology and Medicine in 2025, along with Mary Brunkow and Shimon Sakaguchi, for their discoveries regarding peripheral immune tolerance, gave a historical overview of the eclectic and winding road of the field, up to recent and current clinical testing. Inspired by the valuable information that mutations in humans and mice provided to understanding immunity, Ramsdell, then at a genomics-based company in the early 1990s, and colleagues sought to increase this knowledge by creating their own set of mutations. Anticipating slow progress, they looked for other sources and recognized that the Oak Ridge National Laboratory, created as part of the Manhattan project to build the bomb, had had the foresight to study the effects of radiation on mammals, and had a large radiation-exposed mouse colony, and were looking for induced mutations to study fundamental biology. One mouse strain, Scurfy (which incidentally was a spontaneous mutation, as the founder mouse was not exposed to radiation), had an X-linked disease characterized by massive autoimmune symptoms, spanning various pathologies (diabetes, psoriasis, hyperactive T cells, etc.), suggesting that tolerance had broken down. Back-crossing targeted the mutated region to about 500 kb, a sequenceable region at the time, and Ramsdell set off to sequence the mutated mouse and normal mice to identify single missense mutations that might be responsible. The last gene on his prioritized list of open reading frames, a transcription factor FKH (Forkhead) had a mutation. The homologous gene in humans, Foxp3 (encoding Scurfin), was identified in humans by analyzing families with a rare X-linked immune dysregulation disorder with symptoms similar to the Scurfy mouse. Transgenic expression of the normal Foxp3 gene in Scurfy mice corrected their defective phenotype. Interestingly, when Foxp3 was expressed only in the thymus, symptoms were not corrected, indicating that Foxp3 was involved in peripheral tolerance. The only T cell population that naturally expressed the gene were regulatory T cells (Tregs; identified by Sakaguchi), which were found throughout the body. Transgenic expression of Foxp3 in normal T cells led to hyporesponsiveness, suggesting that this gene could act as a rheostat that could inhibit autoimmune disease, or allow better tumor control, if there was a way to use it. As an undruggable transcription factor, it took 10 years before cell therapies, led by oncology, opened up this opportunity. During this time, enormous information was accumulated regarding the biology of these cells, including the ability to isolate these cells from humans. Multiple companies then were formed to turn Treg cells into a “polypharmacy” due to their many properties aimed at suppressing and repairing uncontrolled immunity. An initial target was rheumatoid arthritis (RA), where fewer Treg cells are present, but still functional, and isolatable from patients. To target these, Ramsdell chose to create a CAR targeting citrullinated autoantigens common in RA using autologous isolated and expanded Treg cells. These CitP-CAR Tregs maintained typical Treg properties. (Parenthetically, expressing Foxp3 into non-Treg T cells created a Treg-like phenotype, between Tregs and effector T cells; this is a therapeutic approach that is also ongoing.) In the first-in-human study in RA in heavily pre-treated patients, at the first dose levels, some reduction in the numbers of swollen and tender joints was observed, ultrasound and immunohistochemistry revealed lower inflammation, and biopsies of the synovial fluid (a challenge in this setting) revealed the presence of infiltrated CitP-CAR Treg cells. No safety signals were identified to date. Overall, Ramsdell is highly encouraged by the ability of Tregs to target and control inflammation, potentially eventually curing these patients.
Reading suggestion:
Unraveling the role of soluble CTLA-4
Decoding T cell circuits and programming immunotherapies - Alexander Marson, University of California, San Francisco, California
Alexander Marson delivered a comprehensive keynote on integrating rapidly advancing technologies to understand how genetics program our cells, and how we can reprogram them using high-throughput CRISPR screens and single-cell genomics to engineer next-generation cell therapies through synthetic biology. To systematically map TCR and antigen interactions at a large scale, Marson’s team engineered specialized vesicles, each coded with a single peptide:MHC target, GFP so the particle can be detected, and an internal circular RNA barcode. A library of these particles can be incubated with polyclonal T cells. The T cells that have a binding event are sorted, and single-cell sequencing is performed to capture the RNA barcode from the particle, the TCR sequence from the T cell, and the corresponding transcriptome to track the T cell phenotype. To achieve this, the team used a landing pad cell line, which allows the utilization of a large pool of plasmids, but integration of only a single plasmid at a single site in the genome. Each landing pad cell only expresses one construct, despite being transduced with the whole array of plasmids. The construct encodes a specific peptide–MHC complex fused to an endosomal recruiting complex required for transport (ESCRT) domain, GFP, and an aptamer. Upon expression, the ESCRT machinery drives the budding of extracellular vesicles coated with the targeted peptide–MHC. A co-encoded circular RNA barcode is captured by the aptamer for downstream identification. For proof-of-concept, particles coated with MART-1 antigen specifically demonstrated robust and highly specific binding to anti-MART-1 TCR+ Jurkat cells, but not with anti-NYESO TCR+ Jurkat cells. The team then investigated primary human PBMCs from an HIV-positive individual who had received a DNA vaccine. A pool of particles with 61 different peptides linked to six different HLA alleles was incubated with CD8+ T cells. Sorting GFP-positive cell–vesicle complexes and performing 10x genomics sequencing revealed the circular RNA barcodes, the paired TCR alpha/beta chains, and the whole cellular transcriptome. This confirmed known TCR–antigen interactions and identified novel TCR–viral antigen pairs. For large-scale TCR–antigen mapping in a NSCLC patient treated with TIL therapy, the researchers conducted pooled cloning of 877 TIL TCRs into TCR-KO Jurket cells using TCRAFT, and screened against a library of 2,700 patient-specific antigens, viral antigens, and tumor-associated antigens on 6 different HLA alleles that would present these peptides. The platform identified specific TCRs recognizing distinct neoantigens and viral antigens (EBV), which were subsequently functionally validated for both target binding and cellular activation using an NFAT-reporter assay. Marson then demonstrated that the platform could be successfully extended to the more complex MHC class II antigens, demonstrating that with a known class II epitope, a pooled screening approach using tiled class II MHC peptides across a larger region of the protein demonstrated the expected epitope specificity. Marson then introduced a translational pivot transforming these vesicles into a targeted, fusogenic drug delivery vehicle by incorporating a mutated VSV glycoprotein for antigen-specific mRNA delivery to T cells. To validate targeted delivery, Marson used MART1 pMHC delivery particles and incubated them with primary CDF5 TCR+ CD8 T cells. These engineered particles successfully delivered functional mRNA ex vivo even to rare antigen-specific primary T cells without any off-target delivery. Marson concluded that this technology can be used to identify TCR-antigen recognition, used as a programmable delivery vector to selectively boost anti-tumor T cells, or deplete autoreactive cells or reprogram them into a tolerogenic state.
Reading suggestion:
Scalable TCR synthesis and screening enable antigen reactivity mapping in vitiligo
Agonistic CD137 (4-1BB) anchored immunotherapy (ANK-203) elicits potent 4-1BBL signaling in vitro and therapeutic responses against established tumors without systemic toxicity in vivo - Robert G. Newman, Ankyra Therapeutics, Cambridge, Massachusetts
CD137 (4-1BB) Is a strong costimulatory molecule that can be targeted with the mAb urelumab (anti-CD137). While anti-CD137 can induce immune activation against cancers, it also causes liver toxicity, and doses that are considered safe are unfortunately ineffective. In recent work, Robby Newman investigated a strategy for anchoring CD137 to reduce systemic toxicity. This strategy, which has been demonstrated with IL-12, involves genetically fusing an alum-binding peptide (ABP) to an immune agonist, which is then phosphorylated to bind tightly to alum particles, creating a stable, long-term therapeutic reservoir that allows for extended tumor retention. Translating this strategy to a full-length antibody, Newman and colleagues genetically fused an alum-binding tail to the bottom of an agonist anti-CD137. The resulting fusion product was shown to maintain binding for both 4-1BB and alum, and a majority of it (85%) was retained on alum after a challenge with excess phosphate and protein. Further, treatment with the anti-CD137–ABP + alum product (ANK-203) induced more robust 4-1BB signaling than the parent antibody, likely due to alum clustering. Similar results were observed in a humanized dual-flanked tumor mouse model, where two intratumoral injections of ANK-203, both given on the same side 1 week apart, enhanced tumor control on both sides, with a portion of mice showing complete responses and resistance to rechallenge. Serum exposure to ANK-203 was low, and effects on liver transaminases were limited, suggesting a tolerable safety profile that supports continued development of anchored agonist antibodies for clinical translation.
Reading suggestion:
A bispecific targeting PD-L1 and 4-1BB shows potential from culture to clinic
Cancer immunotherapy: target, modality, and timing - Owen N. Witte, University of California, Los Angeles, California
Owen Witte began his talk by discussing how the wide genetic variation in the origins of prostate cancer is often ignored by standard-of-care surgery and radiation, followed by adjuvant androgen targeting therapies. While these strategies can lead to minimum-residual disease, patients frequently relapse later with aggressive, treatment-resistant disease, highlighting the need for additional targeted approaches earlier on. To this end, Witte and colleagues investigated a TCR-engineered T cell approach, with secreted prostatic acid phosphatase (sPAP) as the target antigen, as it is expressed almost exclusively in prostate tissues (though it shows some minimal expression in the esophagus). Seven PAP-targeting, HLA-A*02:01-restricted TCRs with medium affinity (10 uM–1 uM; the “goldilocks” zone) were identified from healthy volunteers, but cytotoxicity was very limited. To enhance TCR potency without altering affinity, Witte introduced a “catch-bond interaction” – a modification pioneered by Chris Garcia that causes an allosteric change to prevent disengagement of the receptor and its paired MHC. This type of bond allows the cells to bind in the same way, but requires much more force to disengage, increasing the binding time and efficacy. In vitro and in vivo, this alteration enhanced the potency of PAP-specific TCRs, and recharging experiments showed that T cells could continue to kill new populations of tumor cells, leading to increased tumor cell killing and reduced tumor volumes in mice. Cross-reactivity was also shown to be limited, supporting future clinical development and testing, which is ongoing.
Switching gears, Witte discussed recent efforts to study pairwise dyad formation between cancer cells and T cells, positing that almost all of immunity is due to cell:cell interactions, and so approaches to study this at scale are important. In previously published work, Witte and team fabricated and assembled “pMHC nanovials”, made with PEG-acrylate and gelatin through a microfluidic device, producing an “inner-surface chamber” that is biotinylated to link reagents through streptavidin and capture cells. As the module-based assembly of this system makes it easily adaptable to various reagents, the researchers loaded nanovials with pMHC molecules so that they could be used to isolate cells expressing cognate TCRs. Further, this system is compatible with RNAseq, allowing for analysis of captured cells at the single-cell level. Witte and colleagues then took this technology a step further, adapting the system to create nanovials with slightly larger chambers to capture cellular dyads and study reciprocal signaling. This allowed them to look at, sort, deep sequence, and analyze 10,000+ paired cells after the development of algorithms to deconvolute transcripts from T cells and cancer cells. While Witte describes the current system as “version 1.0”, further optimization and analysis will allow researchers to use this system to identify genes that are upregulated or downregulated in each cell type upon cellular interaction.
Reading suggestion:
Novel binder designs mimic TCR specificity for pMCH-I
Defining T cell receptor repertoires using nanovial-based affinity and functional screening
Checkpoint blockade
Regulation of CTLA-4 and PD-1 blockade immunotherapy by distinct subpopulations of CD4 and CD8 tumor-resident memory T cells - Fathia Mami-Chouaib, INSERM-UMR1186 Gustave Roussy, Villejuif, France
Fathia Mami-Chouaib discussed recent research in human solid tumors, where a high density of tissue-resident CD8+ T cells (TRM), marked by expression CD103 (the integrin ITGAE), was correlated with improved outcomes following treatment with anti-PD-1/PD-L1. In responding patients, TRM cells expressed high levels of PD-1, produced granzyme B, formed stable conjugates with tumor cells, and killed tumor cells, especially upon the addition of anti-PD-1/PD-L1. This cytotoxicity was abrogated with antibody blockade of CD103. As less is known about the role of TRMs in the context of anti-CTLA-4, Mami-Chouaib found that in the MC38 colorectal mouse model, antibody blockade of either CD8 or CD103 did not impact anti-CTLA-4-mediated antitumor responses. However, responses were inhibited by blocking CD4 or CD49a (the integrin ITGA1). Further investigation revealed that tumor control was associated with increased cytotoxic activity of CD4+ TILs towards tumor cells, which was also correlated with increased expression of granzyme B. This cytotoxic activity was inhibited by blocking CD49a. In human melanoma samples treated with dual checkpoint blockade, a high density of CD49a+CD4+ TRM cells correlated with better progression-free survival, and could be used to predict responses. Further, scRNAseq of human melanoma-derived CD49a+CD4+ T cells displayed a specific signature enriched for CTLA-4- and cytotoxicity-related transcripts. Specifically, there was an ITGA1+CD4+ TRM cluster expressing CTLA-4, ITGAE, ICOS, PRF1, GZMB, and transcription factors EOMES and RUNX3. Overall, these results suggest that in NSCLC, an increased density of CD103+CD8+ TRM cells in tumors was associated with improved responses to anti-PD-1, while in melanoma, CD49a+CD4+ TRM cells promoted responses to anti-CTLA-4, and an increased density of these cells was associated with improved responses to combination anti-CTLA-4 plus anti-PD-1.
Reading suggestion:
Different tumour-resident memory T-cell subsets regulate responses to anti-PD-1 and anti-CTLA-4 cancer immunotherapies
LAG-3: The third checkpoint inhibitor and its synergistic interactions with PD-1 - Dario A. Vignali, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania
Lag3 is a well-known immune checkpoint that limits T cell function and disrupts homeostasis. Its canonical target is MHC-II, but according to Dario Vignali, its primary target may actually be TCR signaling. On the T cell surface, Lag3 dimerizes and associates with CD4 and CD8 to disrupt Lck association and T cell activation, interfering with the proximal components of TCR signaling. Adding to the complexity, Lag3 activity depends on ubiquitination for membrane release of the CY domain and inhibitory signaling. Lag3 is also rapidly shed by proteases ADAM10/17, limiting its activity. Importantly, preclinical research and clinical trials have shown that anti-Lag3 and anti-PD-1 act through distinct mechanisms, and combining these treatments can synergize, ultimately leading to the FDA approval of the combination therapy for some indications. To understand what else might couple with anti-Lag3 or the anti-Lag3/anti-PD-1 combination, Vignali highlighted the importance of understanding how Lag3 works, particularly in comparison to PD-1. Comparing adoptively transferred antigen-specific CD8+ T cells lacking both PD-1 and Lag3, Vignali found that they were transcriptionally unique, with a proliferative advantage, reduced expression of Tox, increased IFNγ production, an increased IFNγ response signature, increased tumor infiltration, and enhanced tumor control. Looking more closely at transcriptional changes related to IFNγ production and sensing, Vignali and colleagues found that IFNγ and the IFNγR on T cells were both required for the antitumor efficacy of PD-1/Lag3 KO T cells, suggesting that IFN signaling within T cells may help to sustain their function and/or survival within tumors. Also, in contrast to knockout of PD-1, which had minimal effect on Tox expression, Lag3 knockout substantially reduced Tox. In a clinical trial (HCC 18-071) patients with treatment-naive metastatic melanoma were treated with a lead-in therapy of nivolumab (anti-PD-1), relatlimab (anti-Lag3) or a combination for two treatment cycles (4 weeks), followed by combination treatment for all cohorts. Transcriptional analysis of blood samples taken pre-and post-lead-in therapy showed that among CD8+ T cells, terminally exhausted and pre-exhausted subsets were the primary targets of anti-Lag3 therapy, and showed enhanced TCR signaling in the combination group. These results were validated in patient samples, which particularly showed upregulation of SLP76 phosphorylation. Further analysis revealed that combination-treated groups showed evidence of enhanced cytotoxicity while retaining an exhaustion profile. To understand this apparent paradox, the researchers performed transcription factor network analysis and identified considerable overlap between cytotoxicity and exhaustion programs, with even more coupling of these programs in the combination-treated group. Based on this, and on prior evidence that Lag3 mediates Tox, Vignali suggested that Tox may support longevity and survival of cells, rather than exhaustion. Finally, Vignali discussed the question of whether the lead-in treatment would impact the outcomes of the late combination therapy, and while results are preliminary, there is evidence that patients treated in the lead-in with nivolumab alone responded earlier, but patients treated with lead-in combination therapy eventually caught up. However, lead-in treatment with either monotherapy did appear to reduce outcomes to the eventual combination therapy compared to lead-in with combination therapy. Mechanistically, lead-in with relatlimab alone led to CD8+ T cell clustering with Tregs, which may have contributed to worse PFS outcomes.
Reading suggestion:
Finding the right combination: new insights in anti-PD-1 plus anti-CTLA-4 or anti-LAG3
Antibody-lectin chimeras for glyco-immune checkpoint blockade - Megan J. Priestley, Massachusetts Institute of Technology, Cambridge, Massachusetts
While most cellular recognition research is focused on proteins, Megan Priestley’s research investigates the role of surface glycans, which can be orders of magnitude larger than cells themselves, and are often the first point of molecular recognition between cells. Cancer cells are known to express altered glycans and upregulate sialic acids, which are bound and recognized by one or more members of the Siglec family. Siglecs act as glycoimmune checkpoints, with a mechanism that’s functionally homologous to that of PD-1. While past groups have tried to develop antibodies or decoy receptors targeting this axis, both strategies have advantages and shortcomings. To try to get the best of both worlds, Priestley and team developed an antibody-lectin chimera (AbLec), comprising an antibody-derived antigen-binding domain to a target an antigen expressed on the tumor cell surface along with a glycan-binding domain from a Siglec, with the objective of creating a molecule that binds tightly to and retains specificity for tumor cells expressing the glycan. The model AbLec they developed was specific for HER2 as the target antigen, and utilized Siglec7/9 to target sialic acid. The affinity of this AbLec was nearly as high as that of its parent antibody, trastuzumab (anti-HER-2). Functionally, treatment of HER2+ tumors with this AbLec enhanced macrophage phagocytosis and NK cell- and neutrophil-mediated cytotoxicity against cancer cells, inducing antitumor immunity that was dependent on Siglecs binding to sialic acids. AbLecs also outperformed separate dual blockade of HER2 and Siglec7/9 in mouse models. In humanized metastatic tumor models, AbLec treatment reduced lung metastases better than trastuzumab. An additional advantage of AbLecs is that the antibody base can be easily swapped out or redesigned to target different antigens or even checkpoints, opening up numerous opportunities for dual targeting. Based on these promising preliminary results, Priestley and colleagues are beginning to explore the potential of AbLecs in clinical trials.
Reading suggestion:
Targeting the sialoglycan-Siglec axis for cancer immunotherapy
Cancer vaccination
Leveraging cDC1 populations for enhanced mRNA cancer vaccination - Ross Ward, Icahn School of Medicine at Mount Sinai, New York, New York
Ross Ward highlighted the role of conventional type 1 dendritic cells (cDC1s) and strategies to therapeutically expand them to enhance mRNA cancer vaccines. cDC1s are essential for antigen presentation on MHC class I and class II molecules, effectively cross-priming CD8+ T cells and facilitating CD4+ T cell responses. cDC1s play a crucial role in coordinating complex cellular networks within the tumor immune microenvironment (TIME), and higher cDC1 prevalence in tumors strongly correlates with better clinical outcomes and successful responses to immune checkpoint blockade (ICB). A common therapeutic approach to expand dendritic cells uses Fms-related tyrosine kinase 3 ligand (Flt3L). Flt3L binds to its receptor on dendritic cells and drives downstream JAK/STAT signaling that culminates in cellular survival, proliferation, and differentiation, leading to the expansion of dendritic cells. While recombinant Flt3L therapy has shown synergy with ICB, adoptive cell therapy, oncolytic viral therapies, and DC-targeted vaccines, the short pharmacokinetic half-life of Flt3L (approximately a few hours in mice and roughly 12-24 hours in humans) has hindered its clinical utility. This rapid clearance necessitates challenging, multi-dose regimens (often 4 to 9 daily treatments) to maintain sustained therapeutic levels. Additionally, recombinant Flt3L protein must typically be administered alongside other activating agents or agonists (such as radiotherapy, TLR agonists, or CD40 agonists) to ensure the mobilized DCs achieve proper functionality. To circumvent the limitations of the short protein half-life, Ward and his team hypothesized that an mRNA/LNP delivery platform could provide more sustained and localized production of Flt3L. The intrinsic immunogenicity of the mRNA delivery vehicle itself avoids the need to co-deliver additional exogenous mediators to activate the mobilized DCs. To validate this approach, the team utilized a B16 murine melanoma model. Once tumors reached approximately 3mm in size, mice were treated intratumorally with 3 doses of recombinant Flt3L protein, a single dose of Flt3L-encoding mRNA/LNP, or a control RNA to account for the baseline immunogenicity of the delivery vehicle. Ward demonstrated that a single dose of the Flt3L-mRNA significantly delayed tumor growth compared to Flt3L, establishing mRNA-based delivery as a potent alternative to recombinant protein therapies. Furthermore, intratumoral Flt3L-mRNA/LNP therapy prolonged survival in B16, MC38, and CT26 tumor models, and increased expansion of innate cells (cDCs, pDCs and NK cells [also expanded with the control RNA]) and neoantigen-specific CD8+ T cells in the TIME. In a separate set of experiments, prophylactic mRNA/LNP vaccination with a vector encoding 3 tumor-associated antigens, a B16 neoantigen (Hsf2), and the OVA SIINFEKL epitope (only relevant for in vitro validation) induced antigen-specific CD8+ T cells and protected mice from B16 tumor challenge in a cDC1-dependent manner. Based on these results, Ward and colleagues hypothesized that a combination of intratumoral Flt3L-mRNA/LNP and mRNA vaccination could be used synergistically. Combination treatment with Flt3L-mRNA/LNP and tumor-directed mRNA/LNP vaccination prolonged survival of B16-bearing mice compared to single agents. In summary, intratumoral Flt3L-mRNA/LNP increased recruitment of cDC1, pDC, NK cells, and tumor antigen-specific CD8+ T cells, which synergized with intramuscular tumor antigen-directed mRNA/LNP vaccine that induced antigen-specific CD8+ T cells in a cDC1-dependent manner.
Reading suggestion:
Lipid formulation replacement reroutes mRNA to the spleen for improved immune activation
The T cell precursor frequency determines the immunogenicity of cancer neoantigens - Tzu-Jiun Kuo, Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
Tzu-Jiun Kuo took a T cell-centric perspective to analyze epitopes relevant to cancer neoantigen vaccines within a humanized mouse system. First, sixteen human single-nucleotide variant epitopes – with predicted high binding affinity (except for one) and validated as bona fide epitopes based on detecting T cell responses in patient samples – were selected. Importantly, the wild-type versions of all the epitope regions for the corresponding genes were identical between mouse and human. These were analyzed in a humanized mouse strain (ABab-A2) in which the endogenous HLA and TCR loci were inactivated, and human HLA-A2 and the human TCRαβ loci were inserted. Additionally, a spontaneous tumor (ABab-TUM) that arose in aged mice and had a low tumor mutation burden was used as a relevant tumor model following insertion of a gene cassette encoding the 16 human epitopes. Most tumors were rejected following subcutaneous implantation (some late regression occurred due to antigen-loss variants) and reactivity to the neoantigens was evaluated by IFNγ production following peptide stimulation. Surprisingly, one peptide (#14) dominated the response (with a second being positive, but showing a much weaker response). To understand why peptide #14 was immunodominant, paired alpha and beta TCR chains from T cells specific for peptide #14 were prepared. Sequencing revealed that the CDR3 regions were significantly shorter than those for the naive repertoire, with few N-insertions, suggesting that they were germline-encoded, and thus, that the immunodominance was due to a high precursor frequency, which was validated by tetramer analysis. Interestingly, the variants of the TCR alpha and beta chains for the dominant clonotypes could be shuffled and still retain binding activity, adding to the high precursor frequency. Deletion of #14 and #2 from the cassette resulted in two other peptides showing the strongest responses, supporting a hierarchy of immunodominance.
Reading suggestion:
Location, location, location: why lung tumors trigger weak immune responses in mice
Decoding the determinants of immunogenicity, specificity and escape to a RAS neoantigen - Christopher A. Klebanoff, Memorial Sloan Kettering Cancer Center, New York, New York
Christopher Klebanoff discussed using shared neoantigens not merely as therapeutic targets, but as essential research tools to systematically interrogate and decode the fundamental molecular determinants of immunogenicity, specificity, and immune escape. Highlighting the importance of mutation-derived, HLA-restricted epitopes in mediating human cancer rejection, Klebanoff discussed a seminal clinical proof-of-concept case by Maria Parkhurst of advanced rectal cancer, in which T cells engineered with a single neoantigen-specific TCR cloned from the TILs of a patient led to a durable clinical response that has persisted for years. Despite this clinical validation, Klebanoff emphasized that there are critical gaps in the knowledge to successfully use neoantigen-based therapeutics. Well over 99% of targetable neoantigens are derived from random passenger mutations that are unique to individual patients. Thus, every neoantigen represents an N of 1 experiment, limiting the generalizable principles governing tumor immune recognition. Identifying molecular determinants that dictate whether a naturally processed and presented epitope will successfully incite a spontaneous immune response is critical. Klebanoff argued that identifying these molecular determinants is foundational for optimizing the next generation of treatment strategies – whether to use TILs, engineered TCR-T cell therapies, or personalized vaccines – and also to determine mechanisms of acquired immune resistance. Klebanoff noted that immunogenic point mutations are neither restricted to, nor biased toward the primary HLA anchor residues. In a canonical 9-amino acid peptide, the vast majority of amino acid substitutions occur at non-anchor positions. Consequently, the wild-type counterpart of the mutated epitope retains the capacity to bind the HLA molecule and be presented on healthy tissues, including the thymic epithelium. This shared presentation architecture has implications not only for the safety profile of targeted therapies, but also for the intrinsic quality of the naturally occurring T cell repertoire. To circumvent these limitations, Klebanoff pivoted toward shared public neoantigens, specifically RAS G12 mutations. The pipeline begins with unbiased, mass spectrometry-based immunopeptidomic screening, which identifies two mutation-containing peptides – a 10-mer and a 9-mer at extremely low abundance – as well as a wild-type 10-mer naturally processed and presented on healthy tissues. Both mutant and wild-type 10-mers demonstrate high stability and half-life. In a cohort of 16 diverse patients who shared an HLA-A*11 allele and a somatically acquired RAS G12D mutation, only a minority (3 out of 16 patients) showed a detectable T cell response. In responding patients, brief in vitro stimulation clonally expanded the T cell population that specifically bound to the mutant (G12D) epitope to significant numbers, but not the KRAS WT epitope. To determine the molecular drivers of this immunogenicity, the team compared the cancer genomes of responders versus non-responders. All 13 non-responders maintained diploid cancer genomes, with only a single mutant KRAS allele. In contrast, all 3 responders exhibited profound allelic imbalance, acquiring multiple copies of the mutant allele through copy-neutral loss of heterozygosity or chromosomal amplification. Klebanoff then demonstrated that allelic imbalance increased peptide–HLA site density and was a key determinant of immunogenicity. All three responders also showed LOH at chromosome 6, losing the restricting HLA-A*11 allele as an immune evasion mechanism. In the 50,000-patient MSK-IMPACT cohort, HLA LOH occurred in nearly 25% of all patients, with significant bias toward the loss of the restricting allele in highly immunogenic mutations, like RAS G12V. RAS G12D TCRs retrieved from the responding patients were high-affinity and specific for mutated peptides compared to the wild-type peptide in three-dimensional binding assays. The X-scan analysis confirmed that the retrieved TCRs could only accommodate the aspartic acid substitution at the hotspot position, demonstrating high specificity. In two different preclinical models, single i.v. infusion of RAS (G12D) TCR-T cells mediated durable tumor control. Klebanoff concluded that driver mutations could create actionable public neoantigens, allelic imbalance could initiate spontaneous recognition, and pre-existing immunity could drive HLA-LOH.
Reading suggestion:
Tumoral RNA splicing: source of neoantigens for off-the-shelf therapies?
Cell therapies
IL-12 armored anti-macrophage CAR T cells reset and reprogram the tumor microenvironment to control metastatic ovarian and lung tumor growth - Jaime Mateus-Tique, Icahn School of Medicine, New York, New York
Jaime Mateus-Tique presented a novel therapeutic strategy designed to overcome the physical and immunosuppressive barriers in the tumor immune microenvironment (TIME), which are a major obstacle for CAR T cell efficacy in solid tumors. In ovarian tumors, tumor-associated macrophages (TAMs) infiltrate and surround malignant lesions to form a robust barrier. Single-cell RNA sequencing showed that these TAM populations expressed FOLR2, which strongly overlapped with the immunosuppressive macrophage markers Mrc1 and Arg1. Mateus-Tique and colleagues hypothesized that CAR T cells could be engineered as a "Trojan horse" to selectively target and deliver pro-inflammatory cytokines to these immunosuppressive TAMs, reversing the suppressive phenotype. This TAM-targeting strategy would not be subject to immune escape, as targets on evolving cancer cells are, and allows the CAR T cells to physically anchor within the hostile TIME and locally deliver pro-inflammatory cytokines, reprogramming the localized immune response. To validate this targeted trafficking, the researchers initially engineered an anti-FOLR2 CAR expressing luciferase for longitudinal in vivo tracking. In an ovarian cancer mouse model, systemically transferred control T cells disseminated non-specifically throughout the mice, whereas the targeted anti-FOLR2 CAR T cells successfully homed, localized, and expanded directly within the tumor bed. Transitioning to armed IL-12-secreting anti-FOLR2 CAR T cells (IL-12 FOLR2.CAR T cells), the team encountered severe dose-limiting toxicities. A standard therapeutic dose of 1.5 million cells into tumor-bearing mice led to severe systemic toxicity, driving significant weight loss and early mortality, even in the absence of pre-conditioning lymphodepletion. To mitigate this toxicity, the researchers reduced the infusion dosage to just 100,000 engineered IL-12 FOLR2.CAR T cells and used luciferase-expressing cancer cells to accurately track tumor burden in immunocompetent mice. Low dose IL-12 FOLR2.CAR T with no lymphodepletion reduced tumor burden and extended survival in an aggressive ovarian cancer model. Single-cell transcriptomics and immunohistochemistry revealed that IL-12 FOLR2.CAR reprogrammed the macrophage compartment, increasing CXCL9+ and IL1b+ macrophages and depleting Folr2+ macrophages. Treatment also recruited endogenous T cells into the tumor immune microenvironment (TIME), and these persisted even after CAR T cell contraction. Spatial analysis showed that Gzmb+ CD8+ T cells were in close proximity to the cancer cells, suggesting endogenous CD8+ T cell-mediated tumor control in treated mice. This mechanism was consistent with the observed increase in CD44+IFNγ+ antigen-activated T cells. Turning to changes in the cancer cells, IL-12 FOLR2.CAR T cells upregulated MHC-I and FAS on tumor cells, and FAS deletion in tumor cells impaired immune-mediated cytotoxicity, suggesting that FAS-mediated killing contributed to the observed antitumor efficacy. Mateus-Tique then tested the same approach in a metastatic lung cancer model, targeting a different immunosuppressive macrophage protein, TREM2. Low-dose IL-12 TREM2.CAR T cells reduced tumor growth, increased CD69+CD8+ T cells and MHC-II+ macrophages in the tumor. In summary, IL-12-armored anti-macrophage CAR T cells supported a proinflammatory TIME and controlled tumor growth in an antigen-dependent and FAS-mediated manner.
Reading suggestion:
Breaking bad macrophages through remodeling with armored CAR-T
Reprogramming NK cells to overcome tumor immunosuppression - Rizwan Romee, Dana-Farber Cancer Institute, Boston, Massachusetts
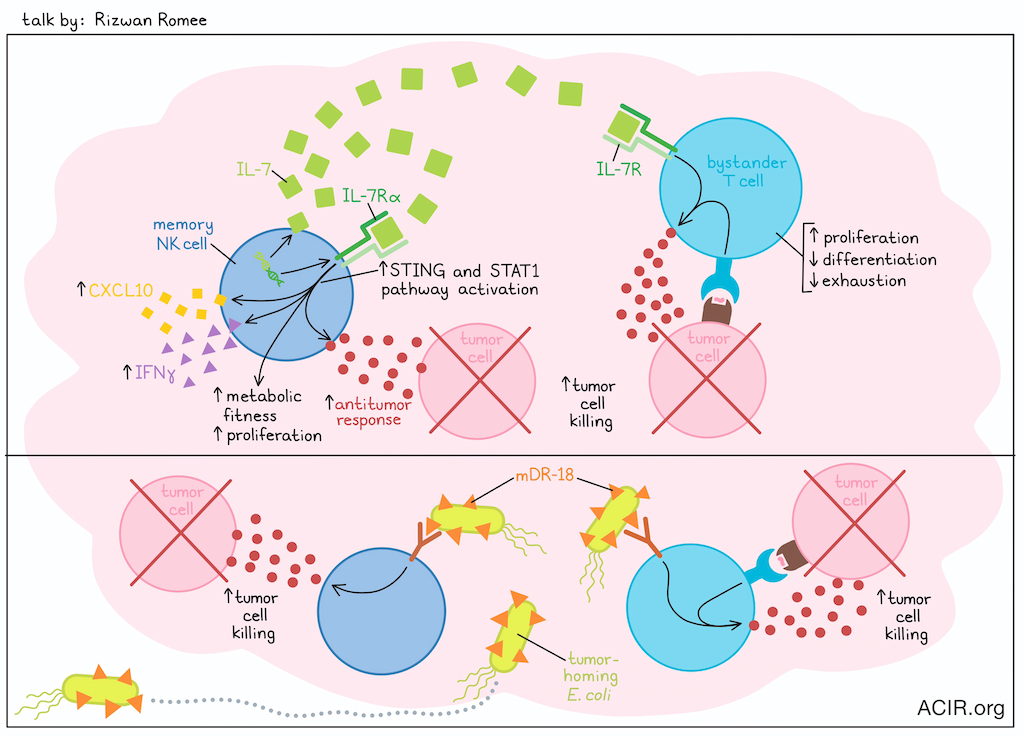
One of the biggest challenges with NK cells for therapeutic use is that they are not long-lived. In past research, Rizwan Romee and colleagues developed memory-like NK cells, which can survive for several months due to their increased expression of ASCT1/2. Using these cells as a platform for genetic manipulation, Romee investigated knock-in of IL-7/IL-7Rα expression to promote NK cell self-activation and enhance NK cell functionality via expression and activation an IL-7/IL-7R circuit in cis (Note: IL-7R is not naturally expressed by most NK cell subsets). IL-7 has multiple beneficial effects on conventional T cells (but not Tregs), including enhanced proliferation and support of an earlier differentiation state and less exhausted state. In NK cells in vitro, IL-7/IL-7Rα expression effectively promoted NK cell proliferation, enhanced metabolic fitness, IFNγ production, and antitumor responses. In vivo, IL-7/IL-7Rα-expressing CAR NK cells showed improved expansion and persistence, and also acted to enhance proliferation and antitumor responses in bystander CD4+ and CD8+ T cells, which naturally express the IL-7R. Treatment also enhanced NK- and T cell-mediated tumor control. Upon further investigation into the mechanism underlying these observations, Romee found that STAT1 and STING pathways drove IL-7/IL-7Rα signaling in NK cells, enhancing their expression of CXCL10 and IFNγ. This mechanism differed from IL-7 pathway signaling in T cells, and is currently under continued investigation. Following their observations regarding the STING pathway, Romee also showed that STING activation could further enhance the cytotoxicity and antitumor efficacy of IL-7/IL-7Rα-expressing CAR NK cells in mice.
Changing directions, Romee discussed the possibility of engineering tumor-homing bacteria to deliver therapeutics and modulate the tumor immune microenvironment (TIME). To this end, Romee’s group utilized a strain of non-pathogenic E. coli and transformed it with plasmids expressing a variety of human or murine cytokines, including IL-15, IL-18, and IL-21. Among these, E. coli expressing an OmpA-tethered decoy-resistant version of IL-18 (E. coli mDR18) had the strongest antitumor effect, leading to improved survival and protection from rechallenge in a portion of tumor-bearing mice, when delivered intratumorally or systemically. This effect was found to be dependent on CD8+ T cells, and to a lesser extent, NK cells, and treatment synergized with anti-PD-1. When E. coli mDR18 was combined with CAR NK cell therapy, Romee saw robust tumor control with enhanced infiltration of CAR NK cells. In ongoing research, Romee showed that bacteria could also be used to deliver bispecific immune cell engagers to further enhance the antitumor immunity of NK or T cells specifically within tumors.
Reading suggestion:
Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia
Neuro-immune interface
From brain to cancer: Harnessing serotonin and antidepressants for immunotherapy - Lili Yang, University of California, Los Angeles, California
To build on the success of immune checkpoint blockade for cancer immunotherapy, Lili Yang evaluated the neuron–immune interface as a potential new source of targets and drugs. Using the B16 model, she evaluated the overlap of molecular pathways in immune, tumor, and neuronal cells, and identified Monoamine-Oxidase A (MAO-A) expression in both neurons and TILs. MAO-A is a key enzyme in the serotonin recycling pathway, and remarkably, the entire recycling network exists in T cells. Serotonin mRNA was upregulated, and serotonin inhibited immunity. Serotonin transporter (Sert) knockout enhanced tumor control, as did treatment with Selective Serotonin Reuptake Inhibitors (SSRIs), a class of drugs widely used to treat depression. Two SSRIs were active across a range of murine models, enhancing T cell activity and synergizing with anti-PD-1. Single-cell RNAseq demonstrated that SSRI-treated and anti-PD-1-treated cell clusters showed little transcriptional overlap. SSRI treatment increased proliferating CD8+ effector cells, mitochondrial activity, and ATP levels, while anti-PD-1 reduced the progenitor exhausted compartment. Antigen stimulation induces serotonin production, which acts as an autocrine factor that restrains T cell activation through a negative feedback loop. Using human PBMCs from healthy donors, Yang showed that Sert and other components of the serotonin recycling pathway were upregulated in CD8+ T cells. SSRI treatment of CD3/CD28-stimulated T cells upregulated cytokine production and TEMRA (effector memory T cells upregulating CD45RA) cells – a major cell type responsible for a beneficial antitumor response – which demonstrated the highest expression levels of Sert and other genes in the pathway. Translationally, in an NSG mouse model bearing NY-ESO-expressing A375 tumors treated with NY-ESO-specific TCR-engineered human T cells, SSRI treatment enhanced tumor control and was associated with T cells producing higher levels of IFNγ and granzyme B. Analysis of TCGA data showed that in patients stratified by Sert expression, lower expression was associated with longer overall survival across multiple tumor histologies. Based on this data identifying the serotonin intratumoral axis as an important and actionable target, Yang and colleagues proposed further retrospective analysis of clinical trial data to identify any potential impact of SSRI co-treatment on outcomes. They are working toward an academic clinical trial evaluating SSRI treatment in neuroendocrine tumors.
Immune microenvironment
Immunology of poorly-immunogenic tumor - Lieping Chen, Yale University, New Haven, Connecticut
In his keynote address, Lieping Chen delivered a mechanistic overview of the biology of non-inflamed, immunologically inactive, “immune-cold” solid tumors. These poorly immunogenic cancers represent over 50% of all human solid tumors, and show minimal infiltration of inflammatory cells, especially T cells. These tumors can be either PD-L1-positive or -negative, and can persist in the presence of systemic T cell immunity. Chen noted that this resistance is often driven by low mutation rates, a lack of innate immune activation, and defective T cell priming, such as that associated with the loss of MHC expression. To identify patients who would benefit from anti-PD1/PDL-1 therapy, Chen conceptually framed the tumor immune microenvironment (TIME) into four distinct classes based on the presence of CD8+ T cells and PD-L1 expression. The first class is double-negative, which lacks PD-L1 and T cells; the second class is double-positive, where both PD-L1 and T cells are present; the third class lacks PD-L1 expression, but has T cells; and the fourth class expresses PD-L1, but lacks T cell infiltration. While the double-positive (class 2) tumors feature active PD-1/PD-L1 interactions and respond robustly to targeted therapy, classes 1 and 4 represent the “cold” tumors that fundamentally lack T cell infiltration. Additionally, class 3 has T cell infiltration, but no PD-L1 expression, suggesting these tumors exploit alternative immune checkpoints to regulate T cell function and evade anti-PD-1 therapy. Across 8 tumor types, the double-negative (cold) tumors accounted for approximately 41% of tumors, whereas class 4 accounted for approximately 11% of tumors. Chen criticized current clinical approaches that empirically apply combination therapies to these non-inflamed cancers, without true mechanistic insight into why the tumor is cold. To determine the reasons behind the lack of T cell infiltration, Chen and colleagues examined the key sequential steps in T cell infiltration: adhesion, transmigration, and migration. Investigating the tumor vasculature, they discovered that an abnormal protein interaction between CD93 and IGFBP7 altered the tumor endothelium, rendering the vessels non-permeable to TILs. Surprisingly, they also found that most of the adhesion molecules on both cancer cells and local blood vessels were not downregulated to evade immunity, but were upregulated. These findings are still under investigation. Turning to T cell mobility and chemokine gradients, the team uncovered no difference in chemokine profiles or their receptors between cold and hot tumors. TCGA analysis of chemokines governing T cell migration revealed three distinct phenotypes: 1) tumors with high chemokines and high CD3+ T cell infiltration, 2) tumors lacking both chemokines and T cells, and 3) a major tumor group characterized by robust chemokine expression, but a lack of T cells across tumor types. Because blood and circulating T cells in this third group expressed normal levels of corresponding chemokine receptors, Chen sought to identify the specific proteins that could antagonize these mobility-enhancing chemokine–chemokine receptor interactions. Using a Boyden chamber migration assay coupled with a genome-wide screen, Chen and colleagues identified Phospholipase A2 Group X (PLA2G10) as the critical hit. PLA2G10, a member of the phospholipase A2 gene family (contains at least 11 members) with both intracellular and extracellular functions, broadly and potently inhibited multiple chemokines on human peripheral T cells. Elevated PLA2G10 expression was associated with reduced CD8+ T cell infiltration in human non-small cell lung cancer. Furthermore, high serum levels of PLA2G10 in advanced NSCLC patients strongly correlated with poor clinical responses to anti-PD-1/L1 therapies. Deletion of catalytic or calcium binding sites reversed PLA2G10-mediated inhibition of T cell mobility. Downstream of PLA2G10, phospholipid metabolites LPC and LPG potently inhibited T cell mobility. Thus, PLA2G10 and its metabolites inhibit chemokine-mediated T cell mobility by suppressing PI3K, but don’t affect proliferation, cytolytic activity, or cytokine production in activated T cells. In addition, PLA2G10-KO mice were resistant to tumor challenge in mouse models, whereas overexpression of PLA2G10 inhibited T cell infiltration and promoted MC38 tumor growth and resistance to anti-PD-1/L1 therapy. Hence, blockade of T cell exclusion factors such as PLAG10 may represent a viable target to enable other immunotherapeutics.
Post-Data World: LLMs and the end of data paralysis - Garry P. Nolan, Stanford University School of Medicine, Stanford, California
Garry P. Nolan delivered a visionary keynote on the transition from an era of "data paralysis" to a "post-data world", urging the integration of Large Language Models (LLMs) to synthesize complex, high-dimensional datasets integrated with the scientific literature. Nolan argued that while bespoke algorithms provide organizational principles for data analysis, they frequently fail to explain the underlying biological causality. We are drowning in data, but unable to connect it to generate meaning. He proposed treating LLMs, trained on corpus of scientific literature, as a panel of experts to extract deeper and actionable meaning, interpretation, and insights, grounded in published knowledge, from your own data. To illustrate, Nolan presented a retrospective analysis of his recent publication. The original study used highly multiplexed proteomic imaging (50-60 markers) and spatial transcriptomics across 78 HNSCC patient lymph node cores. The main finding was that when HNSCC metastasizes to a lymph node, it not only remodels the local metastasis-positive node, but also initiates changes in distal, non-involved lymph nodes, preparing the pre-metastatic niche. Nolan's group identified cancer-associated fibroblast (CAF)-enriched, myeloid-tumor-enriched, and B cell-enriched spatial neighborhoods in patients with poor outcomes. The myeloid–CAF axis obstructed the formation of functional tertiary lymphoid structures, drove perifollicular CD4+ T cell dysfunction, and altered extracellular matrix and chemokine gradients to facilitate immune evasion and further colonization. The conceptual leap occurred when Nolan input the entire manuscript, along with the list of gene and cell types, into an LLM platform called Cellformatica (developed by lab members), generating an exhaustive, fully referenced analytical report. Cellformatica then used Bayesian inference to generate correlations based on other published databases that could be fed into Manus.im (a super-agent; a team of autonomous, linked AI agents), which generated larger causality maps. These maps visually delineated the pathways of immune suppression, distinguishing between relationships directly observed in his lab's data and those inferred from the broader scientific literature. When querying the AI from the perspective of the CAF–tumor interaction, the LLM successfully mapped the downstream cascade, showing that the CAF–myeloid axis drives spatial disruption, leading to specific chemokine alterations (TGFβ and CXCL10), and ultimately resulting in T cell dysfunction and TLS obstruction. Nolan emphasized that this human-in-the-loop approach allows researchers to interrogate their data from multiple cellular perspectives. He concluded that the AI-driven causality maps will soon evolve into predictive agent-based models capable of autonomously identifying therapeutic Achilles heels within complex tissue microenvironments.
Tumor sampling by Myeloid cells - Max Krummel, University of California, San Francisco, California
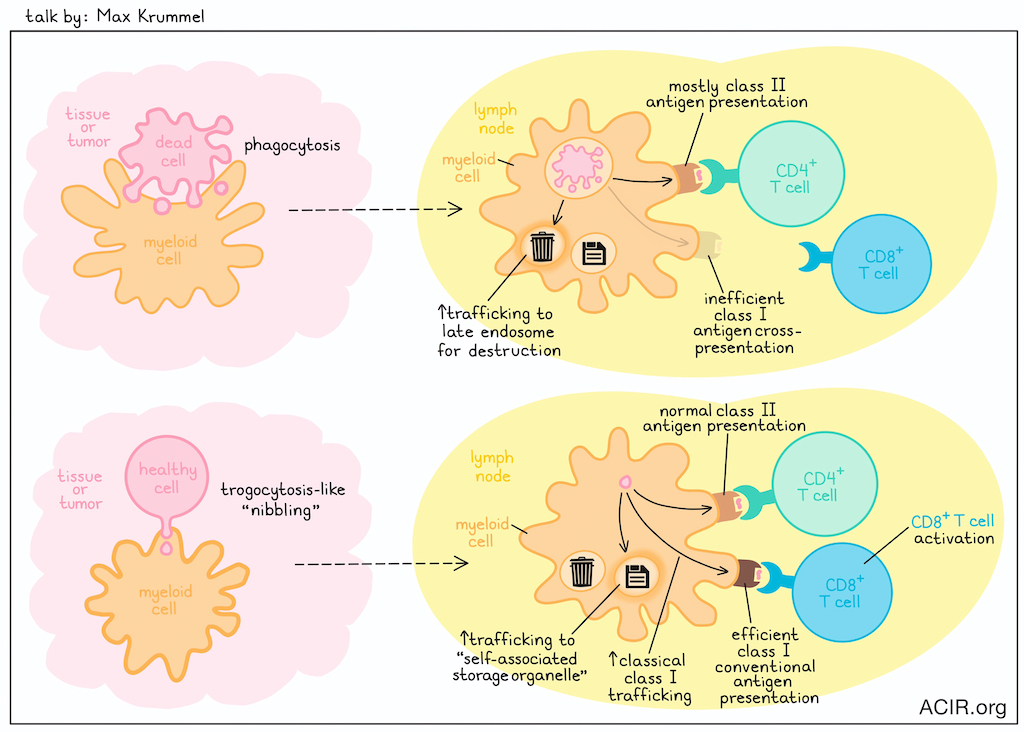
Myeloid cells are known to sample their environment, ostensibly by phagocytosis, to recognize self vs. non-self and present antigens to T cells. However, in this talk,Max Krummel introduced the idea that phagocytosis may not be the only way that myeloid cells sample their environments. This idea was based on observations that myeloid cells at tumor margins were observed carrying small punctates of tumor cytosol, with similar evidence of these cells in draining lymph nodes, which was dependent on CCR7. Video fluorescence observations showed that punctate vesicles could actually be handed off from myeloid cell to myeloid cell. This uptake of cytosol by CD45+ myeloid cells was observed in normal (lung and skin) and tumor tissue (B16). Based on these observations, Krummel investigated whether myeloid cells could sample live cells without destroying them. To this end, he created an assay of bone marrow-derived macrophages (BMDMs) versus healthy cells (mouse embryo fibroblasts). Like in tumors, small amounts of material from healthy cells was picked up by BMDMs, dependent on cell contact rather than cell death. Close-up video imaging showed that upon contact with macrophages, tiny finger-like protrusions on healthy cells were pinched off, releasing small vesicles that were taken up by the BMDMs, consistent with a trogocytosis-like cellular “nibbling” event. These small vesicles were further studied using a small particle spectral flow system, which showed that the ingested material was incorporated into a complex intracellular network for sorting across different vesicular compartments, including some involved in canonical antigen processing. However, unlike with phagocytosis, material ingested through this trogocytosis-like mechanism resulted in less trafficking to the late endosome for destruction, and instead was sequestered in an alternative compartment, that has been temporarily termed a “self-associated storage organelle”. Interestingly, while phagocytosis in BMDMs resulted in endocytosis, with mostly class II antigen presentation and poor class I cross-presentation, live-sampling of cellular material led to classical class I trafficking and efficient conventional class I presentation, maintaining normal levels of class II presentation. Diverting live-ingested material back towards lysosomes reduced CD8+ T cell stimulation. Together, these results suggested that live-sampling may actually be the standard pathway for immune cell sampling of surrounding cells, while phagocytosis is more of an alternative pathway. Further, this mechanism may be responsible for normal immune homeostasis, but may be hijacked by tumors, leading to T cell exhaustion.
Reading suggestion:
Messenger for DCs: vesicles spread tumor antigens across dendritic cell subsets
By Lauren Hitchings, Shishir Pant, and Ed Fritsch



