Predicting TCR recognition from sequence alone is challenging because TCRs with low sequence similarity may bind the same peptide–MHC (pMHC), whereas nearly identical TCR sequences may bind different pMHCs. Wang, Yeh, et al. developed a new approach to map TCR recognition using high-throughput yeast display and protein language models (pLMs). Their work was recently published in Nature Biotechnology.
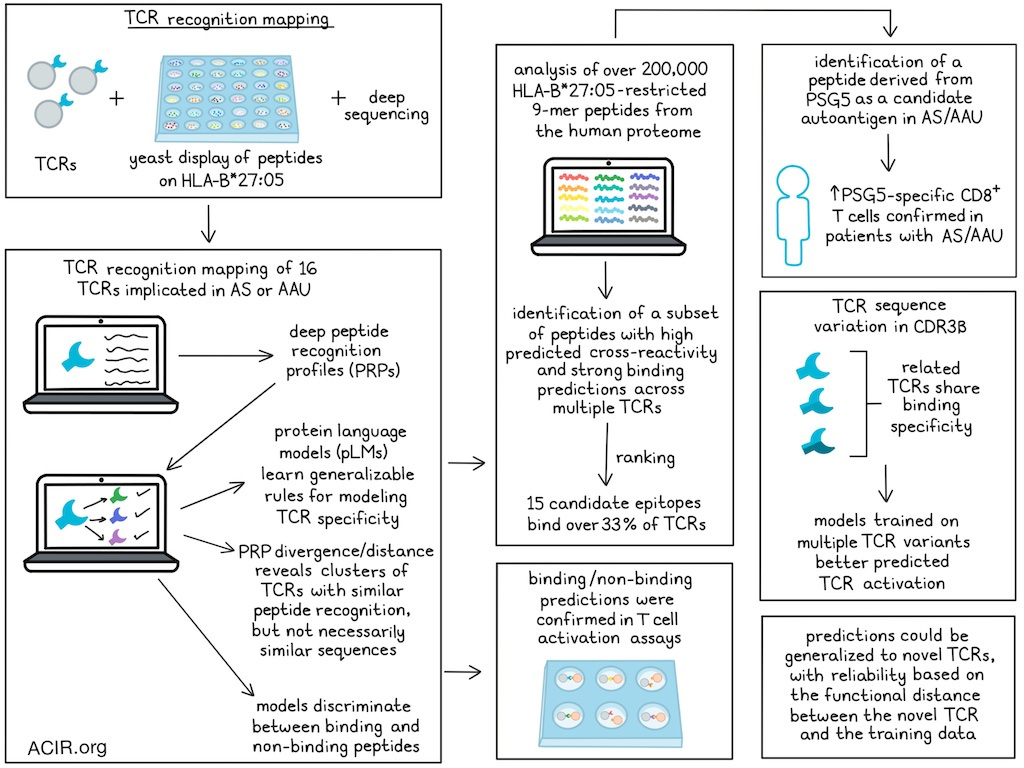
The researchers developed a platform to map the peptide recognition landscape of individual TCRs. High-throughput yeast surface display was coupled with next-generation deep sequencing to quantify binding interactions between a specific TCR and randomized peptide libraries presented on HLA-B*27:05.
The framework was then applied to a panel of 16 HLA-B*27:05-restricted TCRs implicated in the autoimmune diseases ankylosing spondylitis (AS) and acute anterior uveitis (AAU). This generated deep peptide recognition datasets of specific interactions for thousands of unique peptide ligands. These deep peptide recognition profiles (PRPs) provided details of each TCR’s peptide recognition preferences. These data were then used to train pLMs to learn generalizable recognition rules for modeling TCR specificity based on empirical peptide binding. The models were focused on the CDR3β loop, which was previously identified as the dominant mediator of peptide contact in this system. Structural analysis confirmed the focus on the β- vs the α-chain and the predominance of CDR3β contacts.
Alignment of the 16 disease-derived TCRs revealed discrete CDR3 sequence variation within a clonotypically constrained family. The experimental binding landscapes revealed hundreds to >6,000 unique peptide ligands per TCR. The researchers calculated the pairwise Jensen-Shannon (JS) divergence between peptide enrichment probability distributions derived from yeast display data as a functional distance metric reflecting PRP divergence. This revealed distinct clusters of TCRs with similar ligand preferences, largely independent of sequence similarity.
Multidimensional scaling was used to project the TCRs into a peptide recognition space in which JS proximity directly reflected shared peptide specificity. This confirmed that TCRs with similar sequences could exhibit different binding profiles, whereas others formed a tight peptide recognition cluster despite sequence differences.
To use these data in a predictive tool, the researchers fine-tuned pLMs for each TCR to learn its sequence-function map. These models accurately discriminated between binding and non-binding peptides. For 3 of the TCRs, gradient-based saliency methods were used to identify the peptide residues most influential for binding, and amino acid shuffling at each position confirmed the critical residues for accurate binding prediction. These detected residues recapitulated the experimentally derived binding motifs, and aligned with known structurally important contact residues.
The researchers then determined whether the models could generalize beyond the random synthetic peptide library and identify potential autoantigens in AS and AAU. To do so, >200,000 HLA-B*27:05-restricted 9-mer peptides from the human proteome were analyzed, and peptide-binding probabilities for each TCR were computed. This analysis identified a subset of human peptides with high predicted cross-reactivity and strong binding predictions across multiple TCRs. Ranking these peptides by predicted cross-reactivity revealed 15 different candidate native proteome epitopes that could bind >33% of the TCRs.
The researchers then tested whether these binding predictions could identify T cell-activating peptides using T cell activation assays with TCR-transduced SKW-3 cells. This confirmed the ability to discriminate between activating and non-activating peptides, and this discrimination outperformed existing structural modeling metrics. The predicted binding scores correlated with experimental T cell activation across multiple TCRs, including several novel candidate autoantigens. A small subset of peptides activated most of the tested TCRs, suggestive of immunodominant, highly cross-reactive autoantigens in AS and AAU.
Among the newly identified activating peptides, one was derived from PSG5, which is expressed in human iris pigment epithelial cells. In AAU, the iris is a common site of inflammation, suggesting that PSG5 may be a novel candidate autoantigen in AAU. Consistent with this, ex vivo staining with PSG5-HLA-B*27:05 tetramers showed an increase in PSG5-specific CD8+ T cells in patients with AS or AAU.
The researchers then assessed the impact of TCR sequence variation on peptide recognition and model performance. To determine how TCR β-chain diversity influences knowledge transfer to new TCRs, a TCR neighborhood was constructed by engineering one of the 16 TCRs (19.2) with 1-3 amino acid substitutions in its CDR3β region while maintaining the original α-chain. Experimental peptide-binding profiling showed highly similar binding specificities across these engineered TCRs. A pLM trained only on the WT 19.2 PRP data generalized effectively to the individual 19.2 mutants, indicating that these related TCRs share interaction principles.
While the models achieved high accuracy in predicting peptide binding, those trained on individual TCRs showed limited ability to predict T cell activation when tested for cross-reactive peptides. Therefore, the researchers tested whether integrating data from the entire neighborhood could yield a more robust model. Training a model on all 19.2 variants improved performance in predicting which TCRs in this neighborhood induced T cell activation. Several previously identified immunodominant human peptides activated these 19.2 CDR3β mutants.
Wang, Yeh, et al. then examined how to generalize predictions to entirely novel TCRs not encountered during training. They hypothesized that the functional distance between a new TCR and the training data determines predictive success more than sequence similarity. To test this, a leave-one-out TCR cross-validation strategy was applied across the full TCR panel.
Standard TCR sequence similarity metrics failed to predict functional divergence, whereas the predictive value was higher when the functional distance (PRP divergence) between the held-out TCR and the training set was shorter. Therefore, relatedness in peptide recognition patterns, not sequence similarity, is the primary determinant of predictive transferability to new TCRs.
To assess whether the model could provide an intrinsic estimate of its reliability for unseen TCRs, the Mahalanobis distance was used to quantify how far a new TCR is from the distribution of TCRs in the learned joint TCR–peptide embedding space. This metric correlated significantly with experimentally measured functional distance (PRP divergence), suggesting that when the embedding places a new TCR close to familiar training receptors, the motif and activation predictions are more accurate.
The platform described in this work can more accurately predict TCR antigen reactivity than sequence-based assessment. The platform, when properly trained with peptides presented by a given HLA allele, may be used for antigen discovery and to help develop immunotherapies using engineered TCRs.
Write-up by Maartje Wouters, image by Lauren Hitchings



