Pancreatic cancer (PDA) is a highly resistant and difficult-to-treat tumor type, often marked by extensive fibroinflammatory stroma and infiltration of immunosuppressive myeloid cells. These myeloid cells, including macrophages and granulocytes, have been noted to have high expression of CCR1, though the role of this chemokine receptor has not been thoroughly studied. In recent work, Zhang et al. investigated the expression profile and function of CCR1 in the tumor immune microenvironment (TIME) of the PDA and its precursor lesions (PanINs). Their results were recently published in Cancer Immunology Research.
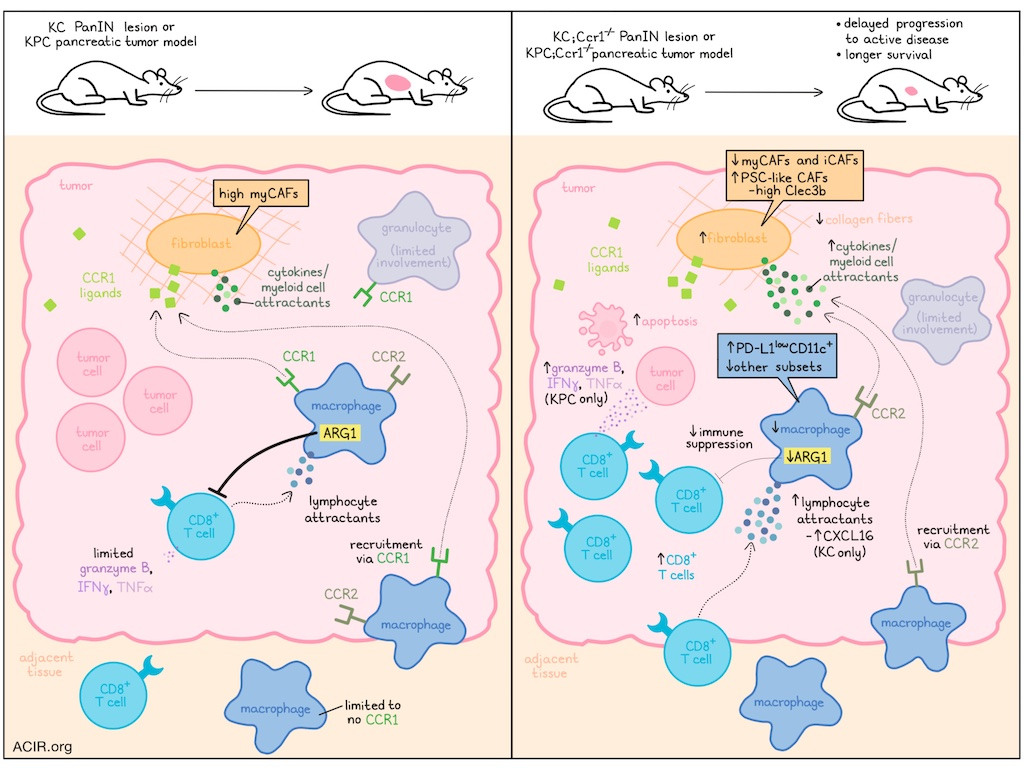
To begin, Zhang et al. evaluated scRNAseq data from human samples of PDA and adjacent normal tissue, and found that CCR1 was the most broadly and highly expressed C-C motif chemokine receptor in tumor myeloid cells, and was particularly expressed in macrophages and granulocytes in tumors, but not in adjacent healthy tissues. CCR2 was also highly expressed in tumor myeloid cells, though it was mostly restricted to classical macrophages, while CCR1 was expressed across TAM populations. CCR1+ TAMs showed increased expression of genes related to polarization, alternative activation, and production of chemokines associated with leukocyte trafficking. CCR1+ TAMs also expressed a higher immunosuppressive signature. Genes encoding CCR1 ligands were highly expressed across a variety of immune cell types in the TIME. Similar results were observed in murine data. CCR1 expression in TAMs was further validated by immunofluorescence and RNAScope in both human and mouse PDA samples.
To study CCR1 expression in vivo, Zhang et al. generated KC mice (KRASG12D mutation introduced into the pancreas to drive PanIN formation) and crossed them with Ccr1-/- mice. In resulting KC;Ccr1-/- mice, which developed normal pancreatic morphology and immune infiltration, the induction of pancreatitis via administration of caerulein induced PanIN formation (as in WT KC mice). Interestingly, while the migration capacity of BMDMs was reduced in KC;Ccr1-/- mice, immune infiltration in PanIN lesions was similar to that observed in WT KC mice, suggesting there may have been mechanisms compensating for the loss of CCR1-induced BMDM recruitment. PanINs in KC;Ccr1-/- mice also showed changes in transcriptional signatures and increased CD8+ T cell infiltration, though granzyme B was rarely detected in either cohort, and mice in both cohorts showed similar lesion burdens, suggesting that the infiltrating CD8+ T cells were not strongly activated.
Given that fibroblasts are a known source of myeloid-recruiting factors, including CCR1 ligands, the researchers evaluated their role in PDA formation and found that in KC;Ccr1-/- tumors, fibroblasts expressed higher levels of cytokines, many of which can act as myeloid cell attractants. The researchers hypothesized that this might help to explain why myeloid cell infiltration remained high in KC;Ccr1-/- lesions, despite the reduction in CCR1-mediated myeloid cell recruitment.
Some of the cytokines upregulated in KC;Ccr1-/- tumors are also known to attract T cells. One such cytokine that was notably increased in KC;Ccr1-/- versus KC lesions was Cxcl16, which was upregulated by TAMs, DCs, epithelial cells, mesothelial cells, and fibroblasts. In an in vitro migration assay, CXCL16 increased migration of CD8+ T cells, suggesting it could be part of a compensatory mechanism for loss of Ccr1.
To study more advanced tumors, the researchers engrafted murine KPC (KrasG12G, Trp53R172H) cells into WT or Ccr1-/- mice and found that in Ccr1-/- mice, CD8+ T cell infiltration and production of granzyme B, IFNγ, and TNFα were increased, apoptosis was increased, and tumor weights were decreased (dependent on CD8+ T cells), suggesting active antitumor immune responses. Similar results were observed with pharmacological inhibition of CCR1 using the CCR1 antagonist J-113863 in WT mice.
Looking more closely at TAMs, the researchers found that PD-L1lowCD11c+ cells increased in tumors in Ccr1-/- mice, while other populations decreased. Further, TAMs showed evidence of reduced immunosuppressive functions, and expressed alternatively activated macrophage markers ARG1 or CD206 in Ccr1-/- mice. Macrophage depletion using anti-CSF1 resulted in decreased tumor weights, demonstrating their role in supporting tumor growth.
Next, the researchers evaluated spontaneous PDA by generating and aging KPC;Ccr1-/- mice. Compared to WT KPC mice, KPC;Ccr1-/- mice showed similar incidence of cancer, but prolonged survival, suggesting delayed progression to active disease. Analysis of primary tumors showed more fibroblasts, fewer macrophages, reduced macrophage expression of ARG1 and proinflammatory cytokines, and increased macrophage expression of Ccr2 in KPC;Ccr1-/- mice. However, the increase in macrophage Cxcl16 expression upon CCR1 loss that was observed in KC mice was not observed in KPC mice.
While fibroblasts do not express CCR1, scRNAseq showed alterations in their transcriptional profiles in CCR1-deficient models. Looking at cancer-associated fibroblast (CAF) populations, Zhang et al. identified 4 distinct subsets - myCAFs, iCAFs, apCAFs, and a novel PSC-like CAF subset with features of pancreatic stellate cells (PSCs) and high expression of Clec3b. While myCAFs were the most abundant subset in KPC tumors, myCAFs and iCAFs were reduced in Ccr1-/- tumors, while PSC-like CAFs expanded. Further, KPC;Ccr1-/- tumors showed reduced CAF activation and production of collagen fibers, providing less support for tumors. Consistent with these results, CLEC3B expression was correlated with longer survival in data for patients with PDA in The Cancer Genome Atlas. Additionally, overall inferred ligand–receptor interactions were strongest between fibroblasts and either epithelial cells or macrophages, but these interactions were reduced in KPC;Ccr1-/- tumors. Two key signalling molecules reduced in macrophages from in KPC;Ccr1-/- tumors were secreted phosphoprotein-1 and thrombospondin-1, which have roles in extracellular matrix function.
Considering possible mechanisms of immunosuppression beyond CCR1, Zhanget al. evaluated blockade of PD-L1, which remained expressed in myeloid cells and other cell types in KPC tumors. In both Ccr1-/- and CCR1 antagonist-treated models, limited synergy was observed with anti-PD-L1. The researchers then hypothesized that because CCR1 ablation reduced expression of Arg1, but maintained expression of Arg2, which could potentially still contribute to immunosuppression. When an ARG inhibitor was added to the combination of CCR1 inhibition and PD-L1 blockade, the triple combination showed the strongest antitumor effect, despite no statistically significant increase in granzyme B-expressing CD8+ T cells.
Overall, these results show that CCR1 is upregulated on myeloid cells in PDA, and plays a suppressive role. Inhibition of this receptor did not affect the formation of disease, but did alter the TIME and slowed the progression of active disease, extending survival in mouse models. Combining CCR1 inhibition with targeting of other suppressive mechanisms improved antitumor efficacy, though there is still room for improvement to better reduce or eliminate disease in this highly resistant tumor setting.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Yaqing Zhang answered our questions.

What was the most surprising finding of this study for you?
Our study yielded many surprising findings. First, while CCR1 was required for macrophage migration in vitro, ablation of CCR1 in vivo did not impair macrophage infiltration, but did impair their immunosuppressive function. Most surprisingly, CCR1 ablation in macrophages resulted in a profound phenotypic shift in cancer-associated fibroblasts. In spontaneous mouse models of pancreatic cancer, CCR1 ablation caused a loss of activated fibroblasts. However, we observed accumulation of a fibroblast population that highly expressed genes associated with pancreatic stellate cells. Thus, ablation of CCR1 might be beneficial through at least two different mechanisms: namely, alleviating immune suppression and driving a microenvironment that is less supportive of tumor growth.
What is the outlook?
Our data indicated that loss of CCR1 reprogrammed tumor-associated macrophages through metabolic regulation – a finding that prompted our current investigation of metabolic pathways and metabolites that may contribute to the immune-suppressive tumor microenvironment of pancreatic cancer. We also found that targeting tumor-associated macrophages by inhibiting CCR1 and Arginase sensitizes pancreatic cancer to anti-PD-L1 treatment in mice. Recent studies have shown the benefit of combination therapies targeting KRAS with mutation-specific or pan-RAS inhibitors alongside immune checkpoint blockade. Our future studies will address whether specific combination therapies might maximize immune responses while reducing unwanted toxicities.
Who or what has been a major source of inspiration or motivation for you throughout your career?
The lack of effective treatments for pancreatic cancer and the high failure rate in translating treatments from mice to humans underscores the critical need to better understand the unique tumor microenvironment of pancreatic cancer and drug resistance mechanisms, which have always been the focus of our research program.



