NK cell antitumor therapies are under development, but the functional states of tumoral NK cells remain poorly understood, limiting progress. Serger et al. profiled NK cells from non-small cell lung cancer (NSCLC) samples, identifying targetable subsets for immunotherapy. Their findings were published in Science Immunology.
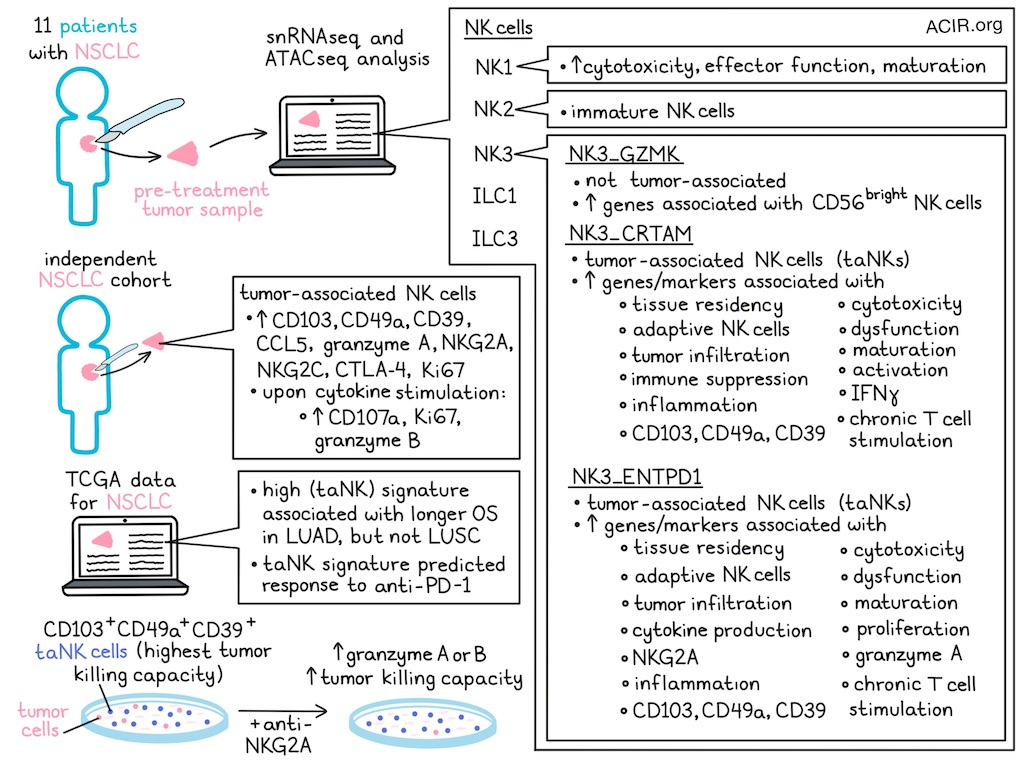
Single-nucleus RNAseq and ATACseq were performed on tumor resections from 11 patients with NSCLC obtained prior to treatment, including lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC). After selecting for NK cells, weighted nearest neighbor analysis was performed to integrate the data. This identified 6 clusters; one cluster only present in 1 patient was excluded. The 5 remaining clusters were annotated based on key marker genes, chromatin accessibility, and publicly available signatures. The identified profiles resembled NK1, NK2, NK3, ILC1, and ILC3 cells. NK3 cells were enriched for a lung tissue-resident NK (trNK) signature and had features of adaptive NK cells.
The NK3 subset could be divided into 3 populations. The NK3_GZMK cluster expressed genes associated with CD56bright NK cells, whereas NK3_CRTAM and NK3_ENTPD1 were enriched for genes linked to tissue residency, adaptive NK cells, and cytotoxicity, and had chromatin accessibility at loci linked to dysfunction and tumor infiltration.
To reconstruct gene regulatory networks (GRNs), the researchers used the multi-omics framework SCENIC+, which integrates transcription factor expression, enhancer accessibility, and target gene co-expression. This revealed that NK1 cells were enriched for cytotoxicity, effector function, and maturation; NK2 cells for immature NK cells; and NK3 cells for NK maturation and tissue residency markers. NK1 and NK2 clusters matched profiles previously detected in peripheral NK cells. Within the NK3 subsets, NK3_CRTAM expressed markers linked to adaptive NK cells and activation. The NK3_ENTPD1 subset expressed markers related to proliferation, cytokine production, and GZMA, while NK3_CRTAM cells expressed IFNG. Both NK3 subsets also exhibited hallmarks of dysfunction and chronic T cell stimulation. NK3_ENTPD1 also expressed high levels of KLRC1 (NKG2A), which is associated with dysfunction in tumoral NK cells.
To identify tumor immune microenvironment (TIME)-specific subtypes, the researchers combined their dataset with two public datasets of non-tumor lung NK cells. They found the NK3 cluster only in tumor samples, and termed these tumor-associated NK3 (taNK3) cells, which highly expressed ITGAE (CD103), ITGA1 (CD49a), and ENTPD1 (CD39).
Compared to non-taNK cells, taNK cells highly expressed genes linked to tissue residency, dysfunction, cytotoxicity, inflammation, and adaptive NK cells. Subclustering NK3 cells revealed three subsets with CRTAM and ENTPD1 marking tumor-associated subsets (taNK), and NK3_GZMK non-taNK cells. SCENIC+ analysis indicated taNK clusters had GRNs related to functional adaptation to the TIME. NK3_CRTAM GRNs were related to immune suppression, maturation, and residency, whereas NK3_ENTPD1 GRNs indicated proliferation, dysfunction, and adaptive responses.
To confirm these findings at the protein level, an independent cohort of NSCLC tumor samples, adjacent lung tissue, and peripheral blood was analyzed by flow cytometry. CD103 and CD49a were among the most differentially expressed genes between taNK and non-taNK cells. Tumor lesions were enriched for CD103+CD49a+ NK cells, and taNK cells showed high expression of CCL5, granzyme A, NKG2A, NKG2C, CD39, and CTLA-4. Projecting taNK cells onto tissue sections using 10x Genomics Xenium spatial transcriptomics revealed co-expression of ITGAE and ITGA1 within individual NK cells near presumably malignant epithelial cells.
Tumor-isolated taNK cells were cultured with IL-12, IL-15, and IL-18. Without stimulation, CD103+CD49a+ taNK cells exhibited higher Ki67 than non-taNK cells. Cytokine stimulation increased CD107a, granzyme B, and Ki67 in taNK cells more than in non-taNK cells.
To provide sufficient numbers of NK subsets for analysis, the researchers developed an in vitro system to model taNK cells from PBMCs of healthy donors. Exposure to IL-15, TGFβ, or irradiated A549 lung tumor cells induced a taNK phenotype marked by CD49a and CD103 expression. The combination of IL-15 and TGFβ induced CD39 within the CD103+CD49a+ subset. These in vitro-generated CD103+CD49a+ NK cells were used in tumor-killing assays, demonstrating enhanced capacity compared with CD103-CD49a- cells.
In TCGA LUAD data, a high taNK signature was linked to better overall survival, but not in LUSC. In NSCLC patients treated with anti-PD-1 therapy, the taNK signature predicted response to ICB.
To determine the potential of taNK cells for cellular therapy, in vitro expansion was tested using dissociated NSCLC tumor samples cultured with IL-15, K562 cells, or both. taNK cells expanded with IL-15 showed the strongest cytotoxicity.
The intratumoral differentiation trajectories of taNK cells were assessed using MultiVelocity, which predicts cell-state transitions based on chromatin accessibility data. A trajectory was observed from NK3_GZMK to NK3_CRTAM or NK3_ENTPD1. Focus was given to the NK3_GZMK-to-NK3_ENTPD1 path, which starts with genes linked to the CD56bright phenotype, then shifts through an interferon response and GZMB induction, leading to the expression of genes associated with dysfunction, tissue residency, adaptive NK cells, and cytotoxicity.
Based on the ENTPD1 expression, the researchers determined whether CD39 could specify a distinct effector state. Spatial transcriptomics revealed colocalization of ITGAE, ITGA1, and ENTPD1 in the lung TIME. taNK cells from tumor lesions were stimulated with IL-12, IL-15, and IL-18. After 5 hours, more CD39+ taNK cells expressed Ki67; after 24 hours, the CD39+ population showed increased expression of granzyme B, Ki67, and CD107a. Using the in vitro taNK model, CD39+CD103+CD49a+, CD39-CD103+CD49a+, and CD103-CD49a- cells were compared for their tumor killing potential. The highest killing capacity was among CD39+CD103+CD49a+ cells.
The researchers hypothesized that KLRC1 (NKG2A) expression may limit taNK cells’ effector capacity. CD39+ taNK cells expressed the highest levels of NKG2A. To assess the effects of NKG2A blockade, patient-derived tumor fragments were treated with anti-NKG2A for 48 hours. Treatment reduced NKG2A expression on both CD39+ and CD39- taNK cells and increased the proportion of CD39+ cells expressing granzyme A or B. In the in vitro taNK model, sorted CD39+ or CD39- CD103+CD49a+ cells were cocultured with target cells with or without anti-NKG2A antibody. Treatment with anti-NKG2A enhanced the killing capacity of both populations, with the most effect on the CD39+ population.
This profiling study identified a taNK cell population, particularly its CD39+ subset, as an important antitumor effector in NSCLC that may respond to anti-NKG2A treatment. If confirmed in other solid tumour types, these findings could inform NK cell-based immunotherapies.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Clara Serger and lead author Alfred Zippelius answered our questions.

What was the most surprising finding of this study for you?
One of the biggest surprises was discovering that the NK cells that appeared most "dysfunctional" at the transcriptional level were not necessarily functionally impaired. We identified a population of CD39⁺ tumor-associated NK cells that expressed markers commonly linked to exhaustion and tissue adaptation, yet they retained remarkable cytotoxic potential. In fact, these cells emerged as the dominant NK cell population capable of killing tumor cells, and responded particularly well to NKG2A blockade. This finding challenged our initial assumptions, and highlights the importance of studying immune cells directly within human tumors, rather than relying solely on conventional markers of dysfunction.
What is the outlook?
Our study provides a framework for understanding how NK cells differentiate and adapt within human lung tumors. Importantly, it identifies CD39⁺ tumor-associated NK cells as a therapeutically actionable population that can be further activated through NKG2A blockade. The next steps are to better understand how these cells develop, what signals maintain their effector function, and how they interact with other immune populations in the tumor microenvironment. More broadly, we hope these findings will contribute to the growing field of NK cell immunotherapy and help develop treatment strategies that complement or enhance existing T cell-based approaches for patients with cancer.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
Prof. Alfred Zippelius: Focus on important questions rather than fashionable ones. Scientific trends come and go, but truly meaningful biological questions remain relevant. Some of the most rewarding discoveries arise from observations that initially seem unexpected or difficult to explain. I would also remind my younger self that science is a team effort. The best projects often emerge from collaborations, open discussions, and the willingness to learn from people with different expertise. Finally, persistence matters: most experiments fail, most hypotheses are incomplete, but curiosity and resilience are often what make the difference in the long run.
What was the coolest thing you’ve learned (about) recently outside of work?
Dr. Clara Serger: Recently, I’ve rediscovered the profound impact of stepping entirely outside my comfort zone through travel. Immersing myself in unfamiliar cultures and connecting with people from diverse backgrounds has a unique way of reframing my priorities. Additionally, I’ve learned how essential dedicated time in nature is for cognitive restoration. It’s more than just a break from the desk, it actively recharges my mind, clarifies my thinking, and creates a kind of creative headspace that a structured workday rarely allows.



