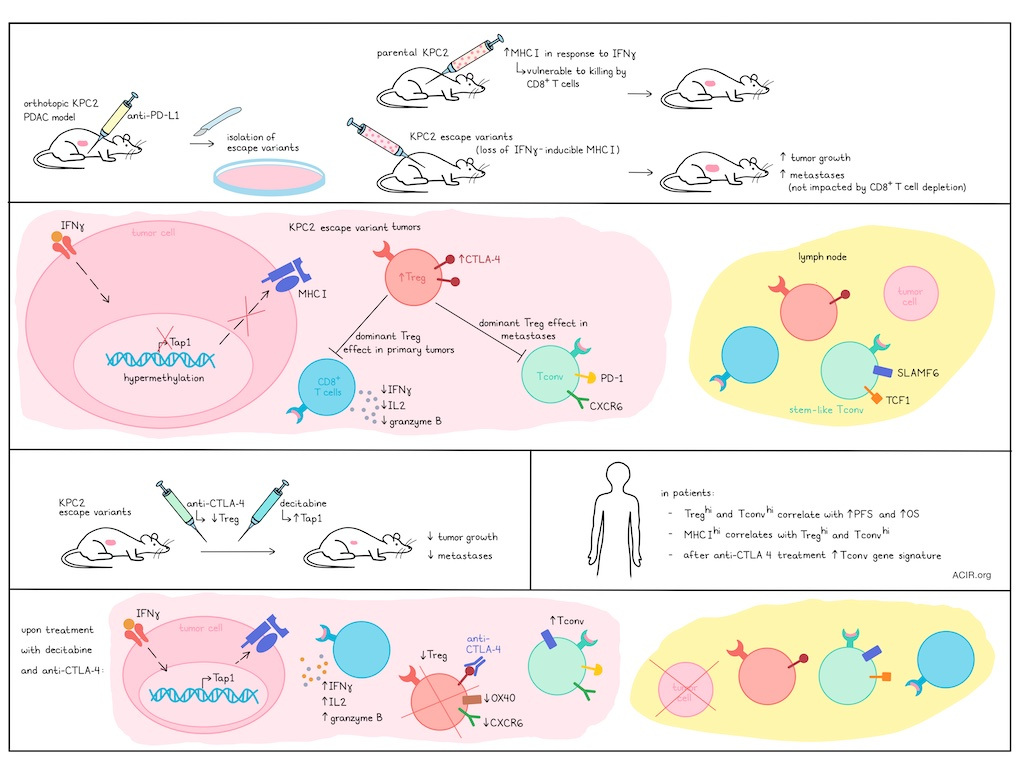
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive and difficult-to-treat-tumor type with complex immune dynamics and multiple immune evasion mechanisms at different stages of disease. In work recently published in Science Immunology, Schmiechen et al. unraveled the roles of IFNγ-inducible MHC-I, CD8+ T cells, Tregs, and conventional CD4+ T cells (Tconv) in promoting or suppressing primary tumor growth and metastasis under conditions of immune escape and/or immunotherapy.
To begin, Schmiechen et al. isolated KPC2 PDAC cells and transduced them with GFP-tagged CBR – an antigen with an immunodominant MHC-I epitope. When the resulting KPC2a tumor cells were implanted orthotopically into mice and treated with anti-PD-L1, the researchers were also able to isolate several escape variants (EscVs). Both parental KPC2a and EscV cells expressed low MHC-I at baseline, but while the KPC2a cells upregulated MHC-I in response to IFNγ, this function was lost in EscVs. In line with this, KPC2a cells were more vulnerable to CD8+ T cell-mediated killing than the EscVs in vitro. In vivo, some EscVs grew at a similar rate as KPC2a parental tumors, but others grew more quickly. Further, EscVs were metastatic, while primary KPC2a cells were not. While CD8+ T cell infiltration was similar between KPC2a and EscVs, CD8+ T cells in EscVs showed reduced signs of exhaustion. Depletion of CD8+ T cells impacted KPC2a, but not EscV tumor growth, suggesting that IFNγ-inducible MHC-I expression was required for CD8+ T cell-mediated antitumor efficacy. This was also observed in a KPC model expressing OVA, suggesting that loss of IFNγ-inducible MHC-I expression may arise as an escape mechanism under strong selective pressure.
Investigating what causes the loss of IFNγ-inducible MHC-I in EscVs, the researchers found that while IFNγ induced similar Stat1 and B2m upregulation in KPC2a and EscVs, expression of Tap1, which transports peptides from the cytoplasm into the ER for MHC-I loading, was upregulated only in KPC2a. Further investigation revealed that while Tap1 and its promoter were not mutated, the promoter was hypermethylated and less accessible in EscVs. Enforced overexpression of Tap1 rescued IFNγ-inducible MHC-I expression in EscVs, reducing their capacity for growth in vivo – an effect mediated by increased proportions of infiltrating antigen-specific, exhausted CD8+ T cells. However, Tap1 overexpression did not prevent EscV metastasis.
To determine the mechanism responsible for the increased metastasis of EscVs, the researchers evaluated other cell types and found that Tregs were increased in EscV2 metastases compared to primary tumors. Treg depletion reduced tumor growth across models, and abrogated EscV2 metastasis, even after tumors were established. Treg depletion also reduced CD8+ T cell exhaustion and increased their production of IFNγ, IL-2, and granzyme B, confirming the suppressive role of Tregs on CD8+ T cells. However, while CD8+ T cell depletion impaired the Treg depletion-mediated antitumor effects in KPC2a, it did not influence tumor growth or abrogate the effects of Treg depletion on metastasis in EscVs.
Turning their attention towards CD4+ T cells, Schmiechen et al. found that co-depletion of Tregs and Tconv abrogated the reduced tumor growth and elimination of metastasis mediated by Treg depletion in EscVs. In KPC2a, co-depletion of Tregs and Tconv promoted metastasis. Deletion of MHC-II on Ccr2+ myeloid cells, which prime Tconv responses, had a similar effect, suggesting that Tregs promote metastasis while Tconv inhibit it. Further, transfer of KPC-specific Tconv into KPC2 tumors induced antitumor and antimetastatic effects. Analysis of Tconv showed that in dLNs, they expressed markers of stemness (SLAMF6 and TCF-1), while in tumors, they reduced expression of stemness markers and instead were more differentiated, with upregulated PD-1 and CXCR6.
In KPC2 tumors, Tregs expressed higher levels of CTLA-4 than Tconv, and in EscV2 tumors and metastases, intratumoral Tregs expressed higher levels of CTLA-4 than Tregs in the dLN and spleen. Treatment with anti-CTLA-4 (clone 9H10 which can mediate Fc-dependent Treg depletion) decreased primary tumors, metastases, and tumor cell dissemination into pancreatic dLNs, dependent on Tconv. Further investigation showed that anti-CTLA-4 partially depleted Tregs and reduced Treg expression of tumoral markers CXCR6 and OX40. Anti-CTLA-4 also increased Tconv in tumors. While Tconv in dLNs were mostly CXCR6−SLAMF6+TCF-1+, representing a stem-like state that can seed Th1 cells after Treg depletion, those in tumors expressed PD-1 (antigen experienced) and CXCR6 (Th1/tissue residency). After treatment with anti-CTLA-4, more PD-1+CXCR6+ cells expressed SLAMF6, representing a more stem-like phenotype. Further, antigen-specific Tconv were almost exclusively CXCR6+PD-1+ in tumors and metastases, but CXCR6-SLAMF6+PD-1low in dLNs, and higher frequencies of tumor-specific Tconv from EscV2-bearing mice were PD-1+ in both dLNs and tumors.
Investigating strategies to overcome the pro-tumor effects of IFNγ-inducible MHC-I loss and Treg-mediated suppression, the researchers evaluated mice with Tap1-overexpressing EscV2 tumors (rescued IFNγ-inducible MHC-I expression) and found that treatment with anti-CTLA-4 reduced intratumoral Tregs while increasing intratumoral PD-1+CXCR6+SLAMF6+ Tconv, resulting in tumor clearance in 40% of mice. Anti-CTLA-4 also eliminated metastatic tumor cells in dLNs and blocked metastasis to peripheral tissues in Tap1-overexpressing EscV2 models, suggesting that combinations of restoring IFNγ-inducible MHC-I, depleting Tregs, and supporting Tconv could hinder tumor dissemination and metastasis. In mice that cleared tumors, tissue-resident tumor-specific T cell frequency was increased in the pancreas compared to the lungs or liver.
Selection against Tap1-expressing tumors was accelerated by anti-CTLA-4 treatment, which also reduced CD8+ T cell exhaustion and increased immunoediting of MHC-I+ tumor cells. Tumor cells in dLNs and metastases were exclusively antigen-negative, suggesting escape through loss of antigen expression. To identify a pharmacological approach to restore MHC-I expression, the researchers tested a panel of drugs and found that treatment of tumor cells with decitabine (a DNA methyltransferase 1 inhibitor) enhanced Tap1 expression and restored IFNγ-inducible MHC-I in both KPC2a and EscVs. In mice, treatment with DNMT1 reduced tumor growth, which was more significant in combination with anti-CTLA-4.
Looking at human data, the researchers found that Tregs and Tconv were positively associated with one another in human PDAC, with higher levels of both associated with longer progression-free and overall survival. Tumors also showed heterogeneous expression of MHC-I, with higher expression positively correlating with Tregs and Tconv, suggesting that these cells may infiltrate in response to higher antigenicity. When the researchers established gene signatures for different CD4+ T cell subsets that expanded after anti-CTLA-4, they found that CD4 Tconv-associated genes were enriched in tumors vs adjacent pancreas tissue, and that this gene signature was associated with longer survival.
These results begin to unravel the complex ways in which IFNγ-inducible MHC-I loss and Tregs contribute to tumor immune escape, while CD8+ T cells and Tconv control primary tumor growth and metastases, respectively. Better understanding these mechanisms enables new and combined strategies to target them, potentially improving immunotherapy in difficult-to-treat cancers like PDAC.
Write-up by Lauren Hitchings, image by Ute Burkhardt.
Meet the researcher
This week, first author Zoe C. Schmiechen and lead author Ingunn M. Stromnes answered our questions.

What was the most surprising finding of this study for you?
ZS: I was most excited initially by the studies over-expressing Tap1 that showed clear rescue of IFN-γ-inducible MHC-I. It’s so interesting that restoring MHC-I upregulation dramatically engaged the CD8 T cell tumor-specific response and cut tumor burden in half yet failed to impair metastasis. Ingunn was also in awe of some of my first experiments after I joined the lab showing complete abrogation of escape variant metastasis after Treg depletion and would often refer back to this finding during our discussions. To see how well MHC-I restoration synergized with CTLA-4 blockade was also incredibly exciting for both of us, really brought the story full circle, and emphasized that both CD8 T cells and CD4 T cells work in concert for the most therapeutic benefit.
What is the outlook?
ZS: I think this study, ongoing work in our lab, and other recent prominent studies on CD4 T cells by other labs, are generating a lot of enthusiasm about the therapeutic potential of tumor-specific CD4 T cells and their role in orchestrating anti-tumor responses. When I joined the lab, we were very focused on CD8 T cells and their dominant role as cytotoxic effector cells, but by the time I finished my PhD, the focus had shifted to include new tools and strategies to manipulate tumor-specific CD4 T cells! There is a great experiment in Figure 4, that shows the anti-metastatic effects of tumor-reactive CD4 T cells by adoptive transfer, and I think it’s a nice teaser for Eddie’s future publications! Overall, engaging CD4 T cells is a fundamental component of effective antitumor immunity and should be incorporated into the design of combination cancer therapies.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
ZS: I chose my graduate school lab based on the people I enjoyed working with and it was the best decision I ever made! I also loved working in the Center for Immunology, which houses world class immunologists and also the nicest and most helpful people. My advice is to get to know as many lab neighbors as possible, you never know what you’ll learn from them, when you might need their help, or if they will become one of your closest friends! Science is a community effort and it’s amazing how much of a difference good relationships make when you’re working long hours in the lab!
Who or what has been a major source of inspiration or motivation for you throughout your career?
IS: Understanding how the immune system works and to ultimately use that knowledge to develop better therapies for patients is a major source of motivation. I believe strongly that through collaboration, following the data, and leveraging new technologies to interrogate antigen-specific T cells, we can make meaningful advances. I'm also deeply grateful to the mentors who guided me, the trainees who continually inspire me, and my family, whose unwavering support has made this journey possible.



