
The ACIR team attended the 23rd CIMT Annual Meeting held on May 11-13, 2026 in Mainz, Germany. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Keynote address
Ira Mellman
Breakthrough lecture
Tyler Jacks
Cancer vaccines
Cornelis Melief
Juliane Walz
Adam Grippin
Tumor immune microenvironment
Anna Obenauf
Marij Welters
Fritiof Åkerström
Daniel Peeper
Innovative Tools
Renata Stripecke
Joanna Turley
Targeting Costimulation
Maria Amann
Ignacio Melero
Michael Lotze
Luc Magré
Immune checkpoint blockade
Myriam Chalabi
Diether Lambrechts
Keynote address
The coming renaissance of cancer immunotherapy - Ira Mellman, Parker Institute for Cancer Immunotherapy and Medici Therapeutics, USA
Ira Mellman delivered his Keynote Lecture promising some science, some editorializing on where the field has been and where it can go, and an introduction to the European IO community of the Parker Institute for Cancer Immunotherapy (PICI), its mission and expansion plans. With the cancer immunity cycle - designed to highlight the key opportunities for productive intervention - as a backdrop, Mellman noted that current approved modalities have been very T cell-centric, focusing mostly on modulating T cell activation (e.g., anti-CTLA-4, IL-2) and blocking exhaustion (e.g., anti-PD[L]-1, anti-Lag-3). However, the search for additional immune checkpoint blockade (ICB) modalities has been mostly frustrating, but Mellman noted that collecting and analyzing human samples from these failures may be more valuable than from successes in terms of future developments. Although still being developed, cancer vaccines have the unique ability to produce new T cells, augmenting the immunity cycle and are beginning to demonstrate clinical activity. Despite the spectacular clinical, and commercial, success of immune checkpoint blockade, the development and approval of other forms of immunotherapy (bispecific T cell engagers, CAR-T cells and TCR-engineered T cells) and the excitement around personal neoantigen cancer vaccines, there are still many deficiencies. Mellman noted that the advent of PD-1/PD-L1 ICB, the biggest factor in the rapidly growing immunotherapy field, was an unexpected success that emerged in the clinic, was embraced by the pharmaceutical industry, but did not reflect long-term thinking or mechanistic understanding. The high bar of this early 'singularity' has not been met again, resulting in waning investment support - and so, this young and successful field needs a renaissance. PICI, which Mellman now leads as President of Research, hopes to contribute to that renaissance.
PICI was founded by serial-entrepreneur Sean Parker, whose understanding of the prospects and problems of applying immunology to cancer led him to envision a new paradigm, focusing on ambitious, not incremental, science by encouraging multi-lab and multi-institutional collaboration, funding game-changing ideas and unlocking researchers’ boldest thoughts. With substantial initial philanthropic funding from Sean Parker, PICI was established in 2016 to accomplish this agenda with a business plan to fund highly collaborative academic science attacking big problems that would fuel start-up biotechs to develop the emergent ideas and a unique value-sharing model that fed funding back to the academic institutions. Multiple major institutions across the U.S. participate and the vast majority of individual U.S. leaders in the field are members. About $65M per year has been disbursed by PICI to fund both academic research at various levels (from institution to young investigators) and new biotechs, and an expansion of PICI funding over the next decade is likely to include EU sites and investigators. Mellman described, with some detail, PICI's priorities for the next decade including attacking solid tumors with T cell therapies, moving beyond CARs to neoantigen (and synthetic neoantigen)-directed therapies with vaccines and engineered TCRs, in vivo cell engineering, systems approaches, particularly understanding the tumor immune microenvironment (TIME), built-for-purpose synthetic modulators and AI support.
Turning to science, Mellman described how parallels in the intracellular phosphatases and targeting elements in the PD-1/PD-L1 pathway have analogies/identities in the TIGIT/PVR/CD155 pathway and, with strong preclinical data, led to intense interest in pursuing TIGIT blockade in the clinic. Randomized phase 2 results for this combination were very promising and led to many phase 3 trials, but none have shown benefit. This lack of effect, Mellman noted, might be due to a number of factors: confounding effects of TIGIT on other cell types, the phase 2 studies having a too small sample size, mouse data not translatable or dual inhibition doubling down on a mechanism that is no longer rate-limiting in patients. The most promising explanation though may be that TIGIT activity depends on acute IFN-I signaling to trigger dendritic cell activity, and in patient tumors, chronic IFN-I signaling may be setting up an IFN-I refractory state, blocking the mechanistic basis for TIGIT activity.
Finally, turning to editorial comments, Mellman outlined the major lessons learned and insights gained from his time in pharmaceutical development. These included: (1) recognizing there are opportunities in the field beyond T cell exhaustion (i.e., beyond anti-PD(L)-1), (2) mice don't predict humans and vice versa, (3) more data is needed on mechanisms of action associated with response, lack of response and resistance, and, (4) most importantly, the most productive areas to focus on are likely to be those with some positive clinical activity but with little R&D. Positive clinical activity is a 'gift from the gods', which should be capitalized on. Two areas of high interest are thus T cell therapy for solid tumors, based on the TIL experience, and cancer vaccines. To support the cancer vaccine focus, Mellman also went into detail describing the recently published and exciting studies in the adjuvant setting of pancreatic ductal adenocarcinoma with a neoantigen-targeted mRNA/Lipoplex vaccine, which together with other studies with mRNA/LNP vaccine in melanoma and a peptide vaccine in renal cell carcinoma, suggest that cancer vaccines may have arrived, at least in the adjuvant setting. (As a research sidelight to these studies, Mellman described a mouse model where a vaccine with a single neoantigen CD4+ T cell epitope was able to induce CD8+ T cell responses and CD8- and cDC1-dependent tumor control in the E0771 model, potentially by inducing epitope spreading within the tumor.) Both cell-based and cancer vaccine approaches are plagued by high cost of goods and long timeframes, but these are engineering and logistical problems that have a higher likelihood of being solved than inventing new biological solutions. Technologies such as microfluidic biochip-based mRNA synthesis, in vivo cell engineering, synthetic TCRs, and synergies between vaccines and cell-based therapies are on the horizon to lead and 'democratize' the renaissance of cancer immunotherapy.
Reading suggestion:
What really happens when you block TIGIT and PD-L1 together
Breakthrough lecture
Probing tumor-immune interactions using genetically engineered mouse models of cancer - Tyler Jacks, Massachusetts Institute of Technology, USA
Recognizing that animal models, particularly mouse models, may not always be directly relevant to human disease, Tyler Jacks used his Breakthrough Lecture to show how these models nevertheless can inform us in important ways about underlying principles, often shared across wide swaths of the eucaryotic kingdom, that need to be investigated in the human setting. Technologies for genetically engineered mouse models (GEMM) have dramatically evolved over the years, to the point where imagination of what to do may now be the rate limiting factor. Jacks focused on how such models can be used to study immune-related detection, interception and disease control. As the first example, the well-described KP GEMM model for lung cancer (conditional activation of mutated KRAS and inactivation of p53) has been shown to recapitulate the normal histological development of human lung cancer, beginning with the cell of origin (alveolar type 2 cells) and progressing to metastatic disease, taking weeks to months to evolve in the mouse. Different from human tumors, disease progression is epigenetically controlled and not due to somatic mutations, so is hidden from the immune system. Prior work showed that adding strong foreign CD8+ antigens made such tumors immune responsive, but only transiently. Taking this approach one step further, Jacks' lab added a known, strong CD4+ T cell epitope, reasoning that this might not only provide important CD4 help in the lymph nodes during priming via IL-2 and/or CD40L, but also could directly interact with the evolving tumor as murine and human alveolar type 2 epithelial cells endogenously express MHC II. Efficient processing of the CD4+ T cell epitope is important, so a cathepsin site was added between the epitope and a membrane-targeting domain. Such tumors generated an immune response against both the CD8+ and CD4+ T cell epitopes, and phenotypically the CD4+ T cells were conventional, Th1-polarized T cells and not regulatory T cells, a balance that depended on the presence of the cathepsin site. Inclusion of the CD4+ T cell epitope enhanced tumor control, leading to longer lived functional CD8+ T cells. To distinguish lymph node priming CD4+ T cell help versus a direct role in the tumor of the MHC II-presented CD4+ T cell epitope, a clever mixing experiment was designed with different colored tumor cells bearing the CD8+ T cell epitope alone versus both epitopes, which clearly showed that the expression of the CD4+ T cell epitope in the tumor immune microenvironment (TIME) was very important. Immunofluorescence demonstrated that having both antigens present increased infiltration of both CD8+ and CD4+ T cells in the tumors with both antigens, and these tumors were highly inflamed. All of these effects were dependent on the expression of MHC II by the alveolar epithelial tumor cells. A vaccine model with one or both epitopes led to similar findings and in particular was able to show enhanced recruitment of functional CD8+ T cells into the tumor only when both epitopes were present, presumably reflecting the more inflamed TIME.
In a second story, Jacks turned to the very difficult to treat pancreatic cancer and its complex highly immunosuppressive microenvironment, typically comprising more non-tumor than tumor cells. Jacks' team speculated, with some supporting data, that the complement system may have a role in the development of such a complex microenvironment. Despite some anti-tumor functions of complement (direct cell lysis and opsonization for macrophage clearance), complement factors C3a and C5a are known to recruit many immune cell types. Complement activation pathways converge on complement factor C3, and a C3-reporter mouse showed dramatic upregulation of C3 expression in KP pancreatic cancer organoids orthotopically transplanted to such mice, primarily coming from inflammatory cancer associated fibroblasts (iCAFs). After showing that such organoids did not develop tumors in C3 deficient mice, Jacks turned to the GEMM KPC model of pancreatic cancer where mutated Kras activation and p53 inactivation occurs only in pancreatic cells. Comparing tumor development in KPC vs KPC/C3-/- mice, KPC/C3-/- survived longer and of those survivors, those mice without grossly detectable tumors (about 60-70% of the animals) had histological lesions reminiscent on the early PanIN stage of PDAC development, with lots of infiltration of CD8+ T cells presumably controlling the further progression of these lesions. Those KPC/C3-/- mice with tumors did not have typical adenocarcinoma, but instead were mesenchymal, undifferentiated, sarcomatoid tumors, a completely different histology. These tumors showed very few CD8+ T cells, suggesting that under immune pressure, tumors of a different histology emerged. Indeed, depleting CD8+ T cells in KPC/C3-/- mice led to the development of adenocarcinomas in all mice. Based on this developing model of C3 induction of C5a and C3a leading to macrophage polarization and a highly suppressive TIME, Jacks showed that inhibitors of murine C3aR or C5aR reduced immunosuppressive macrophage and increased activated CD8+ T cells, and that in an organoid model, these inhibitors improved survival dependent on CD8+ T cells. Combination experiments with other immune and non-immune therapies are ongoing. There are approved inhibitors of C3 and C5aR available, enabling rapid clinical translation.
Reading suggestion:
Hidden gems: cryptic antigens as novel therapeutic targets in pancreatic cancer
When neoantigen expression is low, the T cells won’t go
Cancer vaccines
Benefit from therapeutic HPV vaccine plus PD-1 blocker depends on pre-treatment PD-L1 cancer tissue expression - Cornelis Melief, ISAbella Pharma / Leiden University Medical Center, the Netherlands
With a backdrop of viruses currently being causative of 10-15% of cancers worldwide and the observations that for socio-economic reasons the only two preventative vaccines (one for Human Papilloma Virus (HPV) and one for Hepatitis B virus (HBV)) are not universally utilized and there is an ongoing endemic of HPV-related cancers, particularly among men in the United States and Europe, Kees Melief summarized his past and current work on identifying a therapeutic vaccine for HPV-induced cancers. Although such vaccines can be utilized across various disease stages, from persistent viral infection to recurrent metastatic disease, or in adjuvant or neo-adjuvant settings, Melief emphasized the importance of co-therapy for late stage disease. Melief was the pioneer in the development of synthetic long peptide (SLP) vaccines, which can induce both CD8+ and CD4+ T cells. CD4+ T cells provide help for CD8+ T cells, can directly kill MHC II+ tumor cells and indirectly kill MHC II- tumor cells via induction of tumoricidal macrophage. Further, SLP intercept the antigen presentation pathway much later than other delivery modes which deliver or induce protein synthesis, and are more efficiently processed by DCs. IB101 (peltopepimut-S) is an SLP vaccine comprising overlapping peptides derived from most of the HPV16 E6 and E7 oncogenes divided into two pools for separate injection and formulated with Montanide ISA51. Compared to DNA-based vaccines targeting the same proteins, IB101 yielded 5 - 10-times stronger Elispot results. Based on earlier clinical trials in pre-malignant and late stage disease in combination with anti-PD-1 or chemotherapy which showed durable clinical responses that correlated with vaccine responsiveness, a randomized, double-blind, placebo controlled phase II study in recurrent/metastatic oropharyngeal carcinoma of anti-PD-1 plus placebo (Montanide alone) versus anti-PD-1 in combination with 3 doses of the vaccine was initiated. Treatment started with one dose of vaccine (or placebo) followed one week later by anti-PD-1. The second and third dose of vaccine were administered with anti-PD-1 on days 29 and 50, and then continuing with anti-PD-1 alone. Overall response rates were not statistically different between the arms, however, stratifying patients based on a combined PD-L1 score (CPS) of ≥ 20, the overall response rate doubled or more in the full analysis set and per protocol set (patients who received all 3 vaccine doses). After four years, the median overall survival has not yet been reached for the per protocol set compared to 23 months for the control group, reaching clinical significance. A correlative marker associated with response was neutrophil level. Prior work has shown that HPV16 directly encodes an IL-1 receptor, resulting in significant inflammatory myeloid cell and neutrophil accumulation. Low neutrophil/lymphocyte ratio (NLR) was associated with better overall survival in the entire trial population, and when combined with CPS≥20 was a highly positive prognostic indicator. Melief postulated an overlap between the benefits of an enhanced inflammatory IFNγ signature and an enhanced PD-L1 score.
Reading suggestion:
Vaccine + chemo activate immune response against cervical cancer
Peptide-based Immunotherapy – Bench to Bedside Developments - Juliane Walz University of Tübingen, Germany
Juliane Walz discussed recent developments in peptide-based cancer vaccines. Successful vaccines require the selection of strong peptide targets, which is currently done through two main methods: using algorithm-based prediction based on genomic and transcriptomic sequencing, or using mass-spectrometry-based immunopeptidomics to profile landscapes of HLA-presented peptides. Antigens that could potentially be selected for inclusion in vaccines have typically been tumor-associated antigens or neoantigens, though more recent work has opened up possible targets to include cryptic antigens and inducible antigens, which emerge only in the context of certain treatment regimens. These targets each come with advantages and disadvantages. For example, while tumor-associated antigens may be shared between patients, they may also be expressed in on-target off-tumor tissues, and while neoantigens are highly specific to tumors, they can be challenging to identify and may not always be abundant, as only a small number of mutations result in presented neoepitopes. While neoantigens have been shown to be relevant even in some tumors with low mutation burdens – like in AML, which has identifiable neoantigens that stem from distinct mutation patterns – there are also examples of mutations which do not result in any naturally presented neoepitopes, representing “dark spots” in the immunopeptidome. Recent investigations by Walz and colleagues have identified certain molecular and genetic patterns associated with these dark spots; their work is ongoing. Beyond peptide selection, good peptide-based vaccines need strong adjuvants, and in recent work, Walz and colleagues developed XS15 – a water-soluble TLR1/2 ligand emulsified in Monatinde ISA51VG that allows for persistence of peptides at the vaccination site and induces long-lasting CD4+ and CD8+ T cell responses, with good safety profile. Once peptides and adjuvants have been selected, the next step in vaccine development is to determine a dosing regimen. Walz argued that the adjuvant setting is ideal for peptide vaccination, due to the reduced tumor burden and favorable T cell compartment. However, when patients are treated in the adjuvant setting, they have often been treated with other drugs prior to surgery, so it is important to consider how prior treatments may impact immune landscapes. In one example, Walz described a pre-treatment with a PSMA-targeted bispecific antibody prior to vaccination, and found that it it improved T cell infiltration of tumors, and lysis of PSMA-expressing tumor cells, enabling further T cell priming that was boosted upon vaccination. A randomized phase II trial of this approach in patients with biochemical recurred prostate cancer is planned. In another example, Walz and colleagues studied the use of the demethylating chemotherapy decitabine in the induction of novel neoepitopes by genomic and transcriptomic profiling and identified expression of new transcripts that gave rise to cryptic neoepitopes that could induce antitumor immune responses. Once vaccine peptides, adjuvants, regimens, and combinations are established, the challenge still remains of how to make vaccines available to patients. Walz describes three approaches to this challenge: 1) off-the-shelf vaccines that readily target known antigens, 2) a warehousing approach in which pre-produced peptides can be incorporated into personalized vaccines, and 3) fully personalized vaccines. While these approaches become more costly with more personalization, Walz presented a series of studies showing that each strategy has been proven to be feasible and to induce clinical benefits.
Reading suggestion:
Vaccine with ICB sidekick unlock anti-melanoma T cell immunity
Beyond personalized medicine: A disruptive role for universal RNA therapeutics - Adam Grippin, University of Texas MD Anderson Cancer Center, USA
Personalized mRNA vaccines have shown clinical potential, and may be particularly helpful to induce immune responses in patients without pre-existing immunity and hence do not respond to immune checkpoint blockade, but face significant hurdles, including manufacturing time, complexity, and cost. While working on the development of personalized mRNA vaccines for the treatment of glioblastoma and testing them in companion dogs before starting a small first-in-human trial with 6 patients, Adam Grippin started to also realize that the "linear story" of prescribing which targets to focus the immune system on and building a vaccine for those targets may oversimplify the true complexity of how mRNA vaccines function. In a metastatic osteosarcoma mouse model, Grippin observed that mice treated with a non-tumor antigen (GFP)-targeting mRNA nanoparticle inhibited tumor growth and statistically significantly increased survival time compared to untreated control. This finding got Grippin to explore the use of mRNA therapeutics as universal immune agonists independent of the target antigen that could immediately induce anti-tumor immune responses by relying fully on the innate immune system. Retrospective analysis of patients with lung cancer and melanoma showed that those who received a COVID-19 mRNA vaccine around the time (within 100 days) they began immune checkpoint blockade (ICB) experienced significantly improved survival rates compared to those who did not. This effect was observed across both stage III and IV disease and was not seen for patients treated with chemotherapy and the COVID-19 mRNA vaccine or patients treated with ICB and an influenza or pneumonia vaccine, suggesting a unique synergy with mRNA vaccines. Due to several limitations to this retrospective analysis, a more thorough, statistically designed retrospective analysis was conducted, which confirmed that time to vaccination (within 30 days before initiating ICB in this analysis) correlated with survival advantage, and the advantage persisted across treatment years, and in analysis of patients at another institution. However, other limitations to the analysis existed, including a potential bias towards healthier patients receiving the vaccine. To address this, Grippin and his team synthesized a mRNA vaccine approximating the BNT162b2 COVID-19 mRNA vaccine and observed strong inhibition of B16-F0 and non-small cell lung cancer tumor growth for the combination of ICB and the vaccine. The vaccine induced a surge in IFNα, resulting in systemic immune activation, despite the technological modifications (such as N1-methylpseudouridine) designed to silence TLR7 and minimize IFN production. Although N1-methylpseudouridine was able to eliminate TLR7 activation, the incorporation of N1-methylpseudouridine mRNA with a lipid nanoparticle triggered MDA5 receptor activation, leading to IFNα production and this was abrogated by the knockdown of MDA5. This effect may be due to a high molecular weight RNA species formed during encapsulation, although this observation requires additional investigation. Furthermore, blockage of IFNAR eliminated the added benefit of the COVID vaccine to ICB. Next, Grippin demonstrated that IFNα drove massive activation of myeloid cells, including dendritic cells, macrophages, and inflammatory monocytes. These activated cells present tumor antigens within lymphoid organs, priming anti-tumor T cell responses targeted at multiple tumor associated antigens. Tetramer-positive CD8+ T cells infiltrated the tumor and released IFNγ in response to tumor cells, which in turn led to increased PD-L1 expression on the tumor cells as an immunosuppressive mechanism effectively sensitizing them to ICB therapy. In healthy humans the COVID-19 mRNA vaccine elicited a transient ~100-1000 fold increase of IFNα levels in the peripheral blood and similar human immune cell activation. As a surrogate marker for the induction of tumor-reactive T cells, Grippin and team found increased PD-L1 expression associated with the COVID vaccination on tumors of various histologies. A pragmatic Phase 2/3 multi-institution clinical trial is currently being designed to further evaluate if mRNA vaccines should be integrated into the standard-of-care treatment for lung cancer and melanoma. As the COVID vaccine seems to function as a powerful innate immune stimulant, Grippin is now exploring next-generation immune modulators by modifying the RNA, the nanoparticles, and the encoded proteins, and trying to identify patients most appropriate for treatment to further enhance anti-tumor responses as an off-the-shelf therapeutic.
Reading suggestion:
COVID-19 mRNA vaccines provide a surprising boost to ICB therapy efficacy
Tumor immune microenvironment
Unlocking immunity: Decoding and reprogramming the immune-evasive tumor microenvironment - Anna Obenauf, Research Institute of Molecular Pathology/IMP, Austria
Anna Obenauf combined her interests in targeted therapies and immunotherapy, and investigated the cross-talk between oncogenic signaling and immunity. Of particular interest are the dynamic changes during tumor development and therapy. RAF or MEK inhibitors provide short-term significant benefit but shortly thereafter targeted therapy (TT) resistance develops, and intriguingly patients with TT-resistant tumors respond significantly less well to subsequent immune checkpoint blockade (ICB) or adoptive T cell therapy (ACT) than untreated patients. Modeling TT treatment and development of resistance in mice allowed selection of cell lines from treatment-naive (NTT) and TT-resistant tumors (RTT). Re-implantation of these cell lines and treatment with anti-PD-1/anti-CTLA-4 (or experiments with similar lines from OVA-expressing tumors and treatment with OT-I cells) demonstrated that these tumors were resistant to immunotherapy (IT). Two prospective clinical trials showed that TT before IT resulted in lower overall survival than IT before TT. Interestingly, in vitro RTT cells were sensitive to T cell killing, suggesting that the tumor immune microenvironment (TIME) was altered and responsible for IT resistance. This was confirmed in multiple models by spiking a small number of RTT cells into NTT (IT-sensitive) tumors and vice versa, whereby the TIME from the dominant cell population determined sensitivity or resistance. Detailed imaging of T cell infiltration into tumors revealed that T cells localize initially at the tumor border and co-localize in niches with monocytes and dendritic cells. Single-cell RNAseq revealed dramatic differences at 48 hrs post-ACT among monocytes, DCs (both cDC1s and CCR7+ DCs) and T cells between RTT and NTT tumors (each population being significantly higher in NTT tumors). Immunohistochemistry staining showed localization of most T cells at the tumor border and some of them already in the center for NTT tumors, while all T cells stayed at the border for RTT tumors. Further, high resolution spatial analysis in collaboration with G. Jönsson and J. Boettcher revealed distinct monocyte:T cell and monocyte:T cell:DC interactions in NTT tumors. Functionally, the monocyte:T cell interactions resulted in T cell stimulation (using Nur77GFP+ T cells) and dramatic T cell expansion and infiltration in NTT tumors, with mostly an effector memory phenotype. Using a bilateral tumor model with injection of T cells into only one tumor, Obenauf was able to show that in uninjected NTT tumors, T cells were able to expand, and that T cells injected into NTT tumors were able to transit to RTT tumors, amplify and kill tumor cells, which may contribute to the efficacy of neoadjuvant immunotherapy. In NTT tumors, T cell expansion and tumor control were independent of DCs, and T cell proliferation was linked to the expansion of Ly6a+ inflammatory monocytes. To explain how these inflammatory monocytes were able to present exogenous antigens, a model using tumor and host with different H2 haplotypes demonstrated the ability of these monocytes to acquire antigen:MHC complexes by cross-dressing. Mechanistically, RTT tumors are characterized by higher PGE2 signaling and low IFN-I signaling compared to NTT tumors, and modulation of these parameters shifted the permissive/resistant states, the inflammatory monocyte population levels, and enhanced monocyte:T cell interactions. Retrospective analysis of clinical data has indicated that treatment with NSAIDS (inhibitors of PGE2 production) improved IT responses, which was replicated in preclinical models.
Spatial single cell transcriptomics in oropharyngeal squamous cell carcinoma (OPSCC) - Marij Welters, Leiden University Medical Center, the Netherlands
Marij Welters focused on improving outcomes for patients with oropharyngeal carcinoma, the 6th most common cancer which originates in the back of the oral cavity due to HPV infection and oncogenesis (~70% of cases) or tobacco/alcohol (HPV- ; ~30% of cases). Only a small fraction of patients respond to neoadjuvant immune checkpoint blockade (ICB) and the overall prognosis for HPV- patients is worse than for HPV+ patients. Welters began by summarizing earlier data indicating that among HPV+ patients, those who demonstrated a T cell response (immune response +; IR+) to the viral oncogenes demonstrated significantly better overall survival than those who were IR-, whose survival was close to those of HPV- patients. To understand why some HPV+ patients are IR-, Welters looked at the tumor immune microenvironment (TIME) using CosMx spatial analysis targeting 1000 mRNAs, analyzing the transcriptomes of nearly 4M cells. CosMx also allows antibody staining so immunofluorescence signals can be overlaid. Nine subclusters of tumor cells could be identified, the proportions of which varied between patients indicative of intratumoral heterogeneity. The clusters could be characterized based on HPV signature, state of differentiation, EMT status and hypoxia. Tumor types high in HLA expression were more frequently infiltrated with T cells, suggesting more responsiveness to the HPV antigens. Analysis of the undifferentiated (vs differentiated) clusters demonstrated expression of genes indicative of tumor cell growth. Pseudotime analysis was used to identify cell trajectories, starting with cluster A (high HPV signature), and more differentiated cells were associated with better outcomes, while progression to a less differentiated state was associated with poorer outcomes. These results were validated with TCGA data. Welters then asked whether the evolution of the cancer cells in these HPV+ tumors was linked to other cell types in the TIME. Not unexpectedly, presence of CD4+ or CD8+ T cells and CXCL10+ macrophage were linked to better survival, while certain fibroblast populations or SPP1+ macrophage were linked to worse survival. Advanced clustering of these non-tumor cell types identified 12 core modules, which were characterized by different hazard ratios, with the most favorable module being comprised of favorable immune cells. Spatial cell community analysis showed interaction of cDC1s, favorable macrophage and CD8+ T cells with tumor cell type A (most favorable outcomes). Also present in this most favorable module were cell communities indicative of tertiary lymphoid structures. At the other end, cancer subtype G (poor outcome) showed strong interaction with SPP1+ macrophage. Overall, there was a clear relationship between outcomes and close spatial interactions of tumor cell subtypes with 'good' or 'bad' immune cell neighbors.
Reading suggestion:
CD4+ T cells help cDC1s provide CD8+ T cells with a license to kill
Optimized in situ tumor-to-dendritic cell reprogramming by AT-108 adenoviral vector drives local and systemic antitumor immunity - Fritiof Åkerström, Asgard Therapeutics, Sweden
The cancer immunotherapy approach that Fritiof Åkerström of Asgard Therapeutics presented focuses on reprogramming tumor cells directly within the patient’s body to become cDC1 dendritic cells capable of initiating an anti-tumor immune response. cDC1s are naturally rare, comprising less than 0.1% of a tumor, yet they are crucial for initiating the cancer immunity cycle by cross-presenting tumor antigens to cytotoxic T-cells and their presence is associated with favorable patient prognosis and better responses to immune checkpoint inhibitors. The company’s lead program, AT-108, is a replication-deficient adenoviral vector that delivers transcription factors (PU.1, IRF8, and BATF3) directly into the tumor. Once the tumor cells receive these factors, they are reprogrammed on a transcription and phenotypic level (CD45+HLA-DR+) into cDC1s independent of the starting tumor type. To demonstrate proof-of-concept, Åkerström and colleagues transduced cancer cells in vitro, mixed transduced cells 1:1 with untransduced tumor cells and transplanted them into mice a day later. In a subcutaneous or orthotopic setting in various mouse models, this approach led to complete responses and synergistic effects when combined with anti-PD-1. Furthermore, the reprogrammed cells formed tertiary lymphoid-like structures with BCL6+ germinal centers. Intratumoral injection of Ad5-PIB (AT-108 precursor) into non-T cell inflamed, MHC-Ilow tumors led to complete responses in 50% of the treated animals and survivors were protected against s.c. or i.v. tumor rechallenge. In bilateral tumor models, treating one lesion controlled tumor growth on both sides, suggesting an abscopal effect and a systemic anti-tumor response. Immunophenotyping confirmed that these effects were driven by increased infiltration of effector CD8+ and CD4+ T cells and a reduction in Tregs. The immune memory was supported by the expansion of tumor-reactive and multi-antigen-specific T cells (including neoantigen-specific) within the peripheral blood. Further optimization of the expression cassette of Ad5-PIB refined the therapy, now designated AT-108, maximizing the reprogramming yields in vitro. AT-108 demonstrated improved therapeutic efficacy in preclinical models. Across various patient-derived tumor samples, including melanoma, head and neck, and colorectal cancer, in monolayer format and in organoids, AT-108 reprogrammed tumor cells into cCD1-like cells with upregulated CD141, CD40, CLEC9A and HLA-ABC and increased secretion of pro-inflammatory IFN-β and IL-12p70 in a dose-dependent manner. This resulted in enhanced antigen cross-presentation, allogeneic T cell activation, and autologous tumor cell killing. Back in preclinical models, Åkerström and colleagues tested dosing regimens of AT-108 in combination with anti-PD-1 and anti-CTLA-4 mimicking an intended clinical trial and demonstrated efficacy of AT-108 across different tumor immune microenvironments (TIMEs), including immune-desert, myeloid-rich TIMEs. The research team is currently focused on identifying and validating actionable biomarker assays to monitor payload delivery and expression, as well as downstream systemic immune engagement. Initiation of a first-in-human clinical trial is planned for the second half of 2027.
Reading suggestion:
Flipping the script: reprogramming tumor cells into DCs
Translating the cancer-immune dialogue - Daniel Peeper, Netherlands Cancer Institute, the Netherlands
Daniel Peeper focused his presentation on two primary goals in cancer immunology: understanding the mechanisms of immunotherapy resistance and identifying new targets to increase therapeutic impact. Peeper and his team developed a reductionist CRISPR-Cas9 screening system with a whole-genome CRISPR/Cas9-KO library to identify key mediators of resistance. They applied the standardized system of MART-1-expressing tumor cells being eliminated by MART-1-specific T cells to cell line panels of different tumor origins and observed differential responses that provided a window of opportunity for genetic manipulation to allow or block immunotherapy resistance. Several hits mapped to the TNF pathway. Specifically, knockout of TRAF2 and RNF31 sensitized tumor cells to T cell-mediated apoptosis. Turning their attention to investigating T cells, Peeper and team used CRISPR/Cas9-KO screens to discover that not a single genetic program but distinct genetic circuitries relate intense, acute, or chronic T cell stimulation to T cell dysfunction. While Ctbp1 mediates dysfunction during chronic stimulation and Icam1 during intense and acute stimulation, Dap5 was identified as a central factor involved in all three conditions. Knockout of Dap5 led to increased global translation, reduced cell death, and improved T cell activity and proliferation, enhancing tumor control in adoptive T cell transfer settings. Next, Peeper and his team explored the “dialogue” between tumor cells and T cells. In coculture assays where T cells and tumor cells were labeled with different colors, they identified heterotypic clusters (doublets) formed by the tumor cells and matched T cells. This was observed across multiple histologies. Flow cytometry analysis of tumor digests from melanoma lymph node metastases revealed similar T cell:tumor clusters, and also T cell:APC clusters in all 21 analyzed samples. These clusters comprised an average of 1.4% of the total cells and an average of 12% of the total CD8 T cells were in these clusters. These clusters could be confirmed by multiplex immunohistochemistry and bright field imaging, and were not limited to one T cell per tumor cell and sometimes included all three cell types. Compared to single T cells, T cells from clusters were enriched for tumor-reactive, clonally expanded T cells that exhibit proliferative and exhausted phenotypes, conferring these T cells with a nine-fold higher tumor-killing activity in vitro and in PDX mouse models upon adoptive transfer. Ongoing work is investigating how to incorporate an enrichment module for these cluster-derived T cells into clinical TIL therapy protocols. CRISPR/Cas9-KO screenings for mechanisms of interaction led to the identification of the N-glycosylation pathway as a critical regulator for the T cells/tumor cell interaction, with Mgat2 as a top hit, in two distinct assay set ups. Mgat2 is the rate limiting enzyme in the glycosylation pathway. Knockout of Mgat2 in T cells led to phenotypic changes towards stem-cell-like progenitor states, improving T cell function and tumor cell killing in vitro and in vivo.
Reading suggestion:
Tired out T cells: aberrant glycosylation drives T cell exhaustion in early tumors
Innovative Tools
Facilitating Translation: Preclinical humanized models for immune-oncology research - Renata Stripecke, Cologne University Hospital, Germany
Renata Stripecke began her talk by asking the question - Why do we need humanized mouse models? Most obviously, this is because such preclinical models sit in the drug development cycle between the in vitro drug discovery process and clinical testing, attempting to predict safety, efficacy, and persistence. In immuno-oncology, this can be particularly challenging due to the known plethora of adaptive resistance mechanisms, and the obvious (different MHC alleles and a broad diversity of alleles in humans) and subtle (variations in molecules, pathways and timing) immunological differences from mouse to man. The dramatic toxicity observed in the phase 1 study of the CD28 superagonist TeGenero, which failed to show toxicity in safety studies in mice and non-human primates, further supports the need for better preclinical models, which the FDA has recently identified as a priority, in order to reduce cost and time of IND-enabling toxicology studies, better approximate human cell interactions and to reduce the number of animals required in the successful transition to clinical studies. Crediting Prof. Lenny Schultz for developing the concept of a mouse toolbox that can be used to analyze human cell and tissue samples for translational research, Stripecke briefly charted the history of the development of mouse models deficient in the key immune and myeloid cell compartments (now known as NSG and NRG mice) and emphasized the imperfection of any individual type of model and the importance of optimizing each model for its planned purpose. For immuno-oncology research, key variables include the source of the 'humanizing' cells (fetal liver, cord blood, adult peripheral blood cells), the route of delivery and cytokine support, and the necessary confirmation that the engrafted cells are functioning appropriately. As an example, looking at how CD34+ cord blood cells engraft, Stripecke showed that while female donors generated more consistent CD45+ cell engraftment, male donors generated later CD3+CD45+ T cell development. CD19+ B cell engraftment occurred first, followed by CD4+ then CD8+ T cells and eventually the ability to mount antibody response and reject xenografts. Once matured, such mice can be used in multiple ways. For example, Stripecke's lab developed a system to test antigenicity by ex vivo engineering autologous human monocytes with a lentiviral vector encoding cytokines (GM-CSF and IFN-α) to mature the monocytes to 'induced' dendritic cells and an antigen of choice (e.g., glycoprotein B of human cytomegalo virus) showing that T and B cell responses could be generated, with more advanced systems demonstrating control of viral infection and being capable of generating fully human antibodies with different isotypes and with potential functional relevance. Taking this a step further, lentiviral vectors encoding the common human HLA-A*02:01 and HLA-DRB1*04:01 were co-delivered in vivo with the vaccine vector, and enhanced CD4+ and CD8+ T cell and antibody responses. Stripecke also described a lymphoma model in which Epstein Barr virus is used to develop lymphomas in humanized mice, which can then be treated with CD8+ gp350-targeted CAR T cells and in which CD4+ or total CAR T cells can elicit the cytokine release syndrome. Together these models suggest that multiple problems can be addressed. Finally, Stripecke described mice in which both murine MHC class I and II are knocked out (DKO) to reduce HLA/MHC mis-matching issues, resulting in reduced xenogenic GvHD and hence a longer time period to analyze the activity of adoptive T cells. DKO mice also supported more effective CD4+ (Th1) and CD8+ (cytolytic) T cell development than NSG mice, with higher immune cell expansion and IFN response upregulation upon vaccination. A newer DKO model with human stem cell factor (SCF), GM-CSF, IL-3, and IL-15 support extends the utility of the DKO approach with better engraftment of B and NK cells.
Engineering highly specific peptide: HLA targeting antibodies through epitope guided design - Joanna Turley, Imuno Therapeutics, the Netherlands
The capability to effectively target peptides presented by HLA molecules (pHLA) would significantly expand the potential space of targetable molecules as many more proteins are only found intracellularly than on the surface. pHLA targets are useful for engineered T cells, T cell engagers or antibody-drug conjugates. Joanna Turley focused on the problem of specificity to reduce off-target effects, a problem which has plagued the field. The binding peptide is the basis for selectivity but only represents a very small percentage of the overall pHLA structure and Turley indicated that by carefully evaluating how each peptide docks to the HLA and comparison to other potential cross-reactive species, a better and more narrow list of targets can be identified for selection (and potential cross-reactive species de-selected) during the screening process for antibodies (or TCRs). As examples, Turley showed that the process produced antibodies with high affinity and specificity against three targets (out of three targets that went through the process), which were mostly more selective than other antibodies in development. A specific example is IM810, a T cell engager which binds pHLA from p53R175H, a common p53 mutation presented by the common HLA-A2 allele. The selected antibody binds with high (2.9 nM ) affinity and is cytotoxic to a broad set of cell lines expressing the mutation and the HLA-A2 allele, even some where the level of peptide presentation is below detection by immunopeptidomics. No cytotoxicity is observed if the cell line did not express the mutation. Comprehensive in silico mutagenesis of the peptide and comparison to the native peptidome revealed limited cross-reactivity, and wet lab testing of the potential cross-reactive species showed none were cross-reactive.
Targeting Costimulation
Targeted Costimulatory Agonists: Learning from Forward and Reverse Translation Approaches - Maria Amann, Roche Glycart, Switzerland
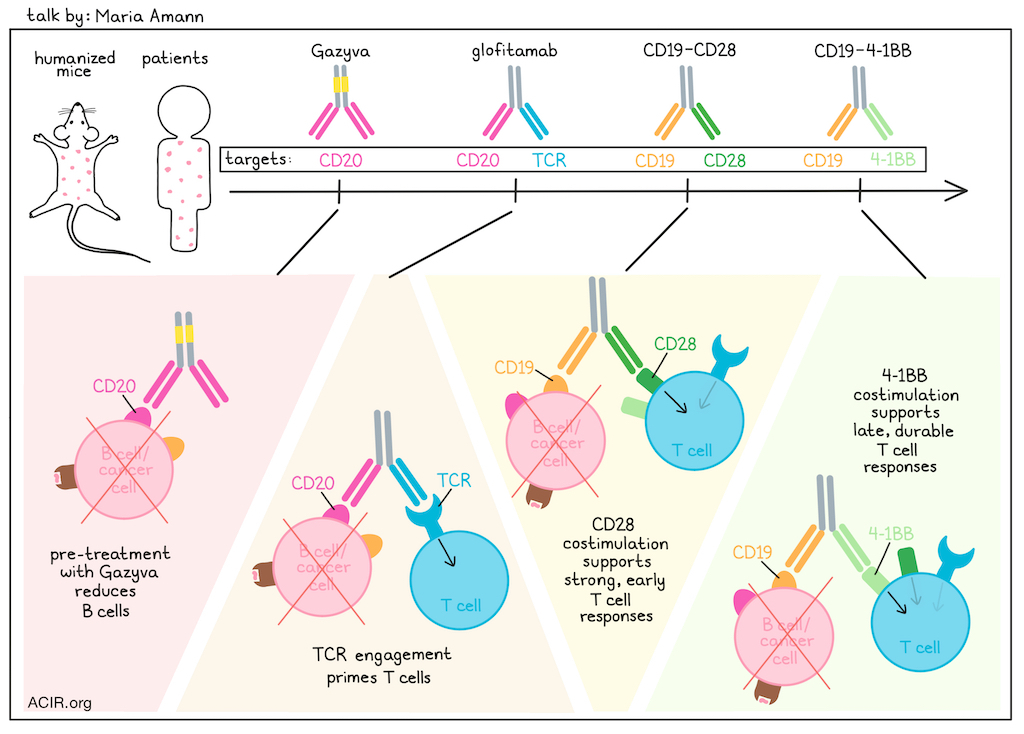
Maria Amann discussed the importance of humanized mouse models to bridge the gap between murine and human systems when evaluating potential T cell bispecific antibodies alone or in combination with costimulatory molecules. One antibody under investigation is glofitamab – a T cell bispecific (TCB) that targets CD3 with one arm (inducing MHC-independent TCR activation), and binds with high affinity to CD20 with the other arm, with a silenced Fc region. In preclinical humanized mice, this bispecific performed fairly well, with response rates of about 40% (CR and ORR), which were similar to those later observed in the clinical setting of Non-Hodgkins lymphoma. To improve responses, the researchers combined glofitamab with bispecifics targeting CD19 and either 4-1BB or CD28 molecules. 4-1BB and CD28 both provide signal 2, but differ in their expression profiles (constitutive vs. activation-induced), and can result in responses with different signal intensity, durability, and toxicity. In a humanized NSG model, Amann and colleagues found that simultaneously administering glofitamab with CD19-CD28 induced substantial cytokine release and weight loss, indicating toxicity. This effect could be mitigated by administering a pre-treatment with Gazyva (a CD20-specific antibody) to reduce B cells, and then administering glofitamab in step-up doses, followed by CD19-CD28. These results translated well from the humanized mouse models to patients, with no incidents of cytokine release syndrome reported in an early clinical trial based on the dosing schedule identified in the mouse. Returning to the comparison of targeting either 4-1BB or CD28, Amann compared their clinical response kinetics and immune responses in combination with glofitamab. The anti-CD19-CD28 bispecific showed strong, early responses and minimal late responses, while the anti-CD19-4-1BB bispecific showed later, but more prolonged and durable responses. The 4-1BB bispecific boosted central and effector memory T cells more effectively. Given these different profiles, Amann tested the triple combination of glofitamab, CD19-CD28, and CD19-4-1BBL, and found that it could achieve long-term tumor control, especially when treatments were administered sequentially. This off-the-shelf combination resembled the effects of 3rd-generation CAR T cells, while offering dosing and scheduling flexibility that could help to manage safety and efficacy. While these studies exemplified how humanized mice can provide relevant data regarding potential combinations, dosing regimens, safety, efficacy, biomarker identification, and more, Amann noted that they do still lack some key translational features, including endogenous priming and tumor antigen-specific T cell biology. To better understand how TCBs would work in a more natural immune environment, Amann and colleagues chronically stimulated and exhausted mouse and human T cells in vitro and showed that they were less reactive to bispecific antibodies. In mouse models, the researchers used B16-OVA tumors to model chronic antigen exposure, and found while there was more endogenous control of tumor growth, there were higher levels of intratumoral exhausted T cells in this setting and the addition of a B16-targeting TCB did not improve tumor control. To model a less intense antigen and in a humanized model, researchers generated a mouse model with physiologically relevant numbers of T cells capable of targeting NY-ESO1. When these mice were engrafted with NY-ESO1-expressing tumors, there was an inherent antitumor immune response which correlated with antitumor response, and evidence of antigen-reactive Tpex cells. When these tumor-bearing mice were treated with a relevant TCB, mice with a higher portion of Tpex cells did not benefit from the TCB treatment, while those with a lower portion of Tpex cells showed improved tumor control. This model was also adaptable, and humanized mice could be generated with different TCR affinities and specificities to better mimic endogenous immunity, which could further enhance the translatability of humanized mouse models prior to clinical testing.
GDF-15 neutralization and CD137 agonists in the immunotherapy clinical arena - Ignacio Melero, Clínica Universidad de Navarra, Spain
Ignacio Melero began by portraying potential combinations of immunotherapies as a painter's palette, with multiple 'colors' to choose from representing ways to increase tumor-reactive T cells, agonist and inhibitory receptors, and molecules to re-model the tumor immune microenvironment (TIME). Moreover, combinations with other therapeutic modalities (chemotherapy, targeted therapies, radiation and surgery) can also be helpful. Additional, 'tools' to inform the best choices for synergy such as biomarkers and preclinical studies are needed. 4-1BB as a target for agonism has multiple favorable properties, primarily that it is found only on T cells that have been stimulated by antigen and is a bona fide signal 2 ligand. Although the signaling pathway is complex, key functional outcomes include inhibition of apoptosis, increased proliferation, cytotoxicity and cytokine production, and genetic deficiencies have some but controllable effects. The first agonistic 4-1BB antibodies taken to the clinic had some clinical activity as monotherapy but also significant liver toxicity, especially for the more active urelumab. To begin exploring combinations, the dose of urelumab was lowered and combination with anti-PD-1 showed a promising response rate in first-line treatment of metastatic melanoma. Preclinical studies with adoptive cell therapy were very successful, demonstrating that T cells in tumors stayed on target longer and killed target cells, which may fit well with 4-1BB+ TIL selection and lends support for the important role of the 4-1BB co-stimulatory domain in CAR T cells. Multiple bispecifics targeting 4-1BB activation to the tumor cell surface are in development and Melero focused on an anti-Fibroblast Activation Protein (FAP) - 4-1BBL bispecific, targeting 4-1BB agonism to fibrotic regions of the stroma. Clinical responses were observed, especially in combination with anti-PD(L)-1, even in immune checkpoint blockade (ICB) refractory patients. 4-1BB agonism (signal 2) was also combined preclinically with a CEA-targeted T cell engager (TCE; signal 1) and demonstrated significant synergy. In the clinic, the safety profile was comparable to that of the TCE, however more activated peripheral T cells, greater infiltration of T cells into tumors and clinical activity was observed for the combination. The newest 4-1BB bispecific (GEN1046) targets agonism to PD-L1 expressing cells and simultaneously blocks PD-1:PD-L1 interactions. GEN1046 showed an acceptable safety profile but activity was dose-dependent due to the need to properly cluster 4-1BB. Activity was recently shown to be due to ligation of PD-L1 on cDC1s and co-stimulation of 4-1BB on T cells, supporting synergy with anti-PD-1 and Flt3L-Ig, which was demonstrated preclinically. In the second part of his talk, Melero turned to describing the newest clinical results with a GDF-15 blocking antibody. GDF-15 is a pleiotropic cytokine with activities in pregnancy (tolerance), prevention of tissue injury, anorexia and multiple aspects of cancer (cachexia, proliferation and metastasis, and immune escape). GDF-15 binds to the active conformation of LFA-1 integrin on T cells and blocks T cell infiltration. It is overexpressed by many tumor cells and has tumor cell intrinsic effects (proliferation and migration) and extrinsic effects. Preclinical models supported anti-tumor activity of antibodies blocking GDF-15 activity alone or in combination with anti-PD-1, and the dose-finding clinical study of an antibody blocking GDF-15 in combination with anti-PD-1 demonstrated safety, deep and durable clinical responses, and an influx of CD8+ and CD4+ T cells into the tumor induced by monotherapy alone. Multiple Phase 2a studies are underway and Melero focused on two diseases, urothelial bladder cancer and lung adenocarcinoma, due to a strong negative correlation between GDF-15 expression and key signatures of immune activity, suggesting these are highly suppressed by GDF-15. In heavily pre-treated or ICB-refractory patients, deep and durable responses were observed in both settings, and also in hepatocellular carcinoma. Intratumorally, the levels of T cells correlated negatively with GDF-15 serum levels and GDF-15 blockade led to increases in IFNγ. Neither soluble GDF-15 levels, CD8+ infiltration levels nor PD-L1 histologic score were predictive markers for clinical activity. To close, Melero described a recent neoadjuvant trial in muscle-invasive bladder cancer comparing anti-PD-1 alone to anti-PD-1 plus anti-GDF-15. Impressively, the combination arm showed a 3-fold increase in histological (both pCR and MPR w/o pCR) and radiological (CR and PR) responses, abrogating the need for surgery in some patients. A separate anti-GFD-15 antibody has also been shown to reduce cachexia in cancer patients.
Reading suggestion:
A bispecific targeting PD-L1 and 4-1BB shows potential from culture to clinic
Signal 2 in TIL Therapy for Cancer - Michael Lotze, University of Pittsburgh Cancer Institute, USA
Michael Lotze, one of the early pioneers in the field of TIL therapy, gave an overview of the field leading to approval in 2024. He covered the impact of targeting signal 2 (co-stimulation) on TIL phenotype, and recent data on some molecules identified by CRISPR KO screening. Canonically, CD28 co-stimulation enhances T cell numbers and function in various important ways, and multiple immunotherapies aim to overcome natural restrictions to CD28 signaling (immune checkpoint blockade, small molecules) or to capitalize on such signaling (CAR T cells, co-stimulatory bispecific antibodies). The development and now approval of TIL therapy, arguably one of the most successful immunotherapies - when it can be used - is however still met with challenges, such as terminal exhaustion of T cells, the suppressive tumor immune microenvironment (TIME) of solid tumors and tumor antigen loss/ immune escape. Augmenting CD28 co-stimulation may provide one approach to enhance T cell number or phenotype. TIL therapy has been shown to be more effective and durable than anti-CTLA-4 therapy, which in principle works through CD28 signaling, so this may be a fertile field for further TIL therapy improvements. The intracellular E3 ligase Cbl-b is a key negative regulator of CD28 and TCR signaling, down-modulating NFAT and NF-κB pathways. Phosphorylation stimulated by CD28 signaling converts Cbl-b to the active state, which downregulates immune responses, and a small molecule (NX-0255) locks Cbl-b into the inactive conformation, enhancing the immune response. In preclinical experiments NX-0255 enhanced IL-2 and IFNγ secretion of primary human T cells upon TCR stimulation, and improved control of murine tumors following adoptive transfer of T cells that have been treated with IL-2 and NX-0255 ex vivo. In a non-clinical study with patient TILs, treatment with NX-0255 (producing drug-enhanced TILs [DeTILs]) increased TIL expansion, TCR diversity and improved multiple aspects of the TIL phenotype. When applied to the tumor fragment culture system developed by Daniela Thommen, activity of DeTILs was high and enhanced by addition of NX-0255 to the fragment co-culture. Multiple cytokines, especially CXCL10, were induced. In a search for new targets to improve TIL therapy, Lotze described a genome-wide CRISPR knockout screen with a readout of intratumoral T cell accumulation. In addition to PD-1 (but interestingly not LAG-3, TIGIT or TIM-3), SOCS1 and REGNASE-1 were the top hits in a single editing screen and contained in the top combos in a dual-edited screen, where Roquin-1 and IκBα emerged as top synergistic molecules. REGNASE-1 is involved in control of mRNA degradation, and Roquin-1 is both an E3 ligase and binds mRNA to promote degradation. Inhibition of SOCS1 and REGNASE-1, which target complementary pathways regulating IL-2 and TCR signaling respectively, emerged as the top combination in preclinical studies. Clinical drug candidates KSQ-001EX (targeting SOCS1) or KSQ-004EX (targeting both SOCS1 and REGNASE-1) were used to enhance TILs during ex vivo culture (eTILs) and demonstrated enhanced IFNγ production, enhanced tumor killing and improved activity during chronic stimulation. A novel tumor-reactive T cell classifier (TRACE) showed no negative impact on the frequency of tumor-reactive clonotypes. In a first-in-human Phase I study, KSQ-001EX was shown to have a tolerable safety profile (w/ and w/o IL-2), 100% tumor reductions above a median dose (independent of IL-2) and an increase in circulating IFNγ, indicative of inhibition of SOCS1. In a case study of a patient with stable disease for over 1 year, persistence of KSQ-001EX eTIL in the circulation and tumor infiltration at day 28 were observed.
A novel PD-1 targeted IL-12 mutein construct to specifically activate intratumoral T cells - Luc Magré, Erasmus MC, the Netherlands
To safely utilize cytokines to remodel the tumor immune microenvironment (TIME), strengthen the activity of intratumoral lymphocytes and myeloid cells and enhance the activity of immune checkpoint blockade, Luc Magré constructed a PD-1-targeted version of IL-12. IL-12 is potent cytokine boosting Th1 cell activity and IFNγ production, but systemic toxic effects, particularly on NK cells, have limited use. Magré reasoned that targeting IL-12 via a PD-1-directed antibody would focus activity to PD-1-expressing T cells in the TIME and limit systemic effects. Furthermore, a mutein of IL-12 (IL-12m) was utilized with reduced receptor affinity to restrict binding only when assisted by the cis interaction of the anti-PD-1 binding, reducing systemic interactions. In a murine MC38 model resistant to anti-PD-1 therapy, anti-PD-1:IL-12m controlled tumor growth as well as isotype:IL-12 but without the systemic side effects. To demonstrate activity in a human system, Magré first showed PD-1 upregulation on CD4+ and CD8+ TILs (from CRC and HCC samples) but not NK cells, and then demonstrated STAT4 phosphorylation (the proximal signal after binding to the IL-12 receptor) and downstream IFNγ and granzyme B upregulation in the human samples, mostly dependent on the anti-PD-1 targeting domain on the immunocytokine and PD-1 expression on the T cells. TCR sequencing revealed expansion of the most abundant clonotypes in the two patients analyzed. Utilizing the tumor fragment culture approach developed by Daniella Thommen, following anti-PD-1:IL-12m stimulation, although heterogeneity in the expressed immune markers was observed, IFNγ secretion could be used to distinguish sensitive and resistant cells. Other biomarkers for potential responsiveness included a negative correlation with presence of Treg cells and a positive correlation with CD39+ CD8+ T cells.
Immune checkpoint blockade
Breaking barriers with neoadjuvant immunotherapy - Myriam Chalabi, Netherlands Cancer Institute, the Netherlands
Myriam Chalabi began her presentation by highlighting the evolving role of neoadjuvant immunotherapy in cancer treatment and emphasizing its advantages over adjuvant therapy by inducing much broader and more clonal T cell responses. She also stressed its potential for practice-changing clinical results by citing a recent trial showing that patients with advanced melanoma treated with two cycles of neoadjuvant immunotherapy exhibited longer event-free survival compared to those receiving a full year of adjuvant treatment. Beginning with the "NICHE" window of opportunity trial in 2016, Chalabi has been studying the treatment of patients with mismatched repair-deficient (dMMR) and -proficient (pMMR) colorectal tumors with two cycles of anti-PD-1/anti-CTLA-4 neoadjuvant immunotherapy. 98% of patients with dMMR colorectal tumors responded, with 95% and 68% achieving a major and complete pathologic response (mPR and pCR), respectively. The pathological response rate observed with standard neoadjuvant chemotherapy is 7%. While the vast majority of patients had circulating tumor DNA (ctDNA) present at the start of therapy, 83% and 100% of patients were free of ctDNA before surgery and after surgery, respectively, which is in line with all patients being disease-free after 3 years. Interestingly, β2M wildtype and β2M mutant tumors responded equally well to treatment and research suggested that γδT-cells play a role in the response to immunotherapy in these cases. Another trial, in which 59 patients were treated with anti-PD-1/anti-LAG3 demonstrated similar encouraging results with a 97% mPR and 69% pCR. Unlike dMMR tumors, MMR-proficient tumors are typically "cold" and non-immunogenic. Anti-PD-1/anti-CTLA-4 neoadjuvant immunotherapy led to complete or near complete responses in 8 out of 31 pMMR patients in the NICHE study, a notable finding given the poor response to the same combination therapy in the metastatic settings. Responsiveness in pMMR tumors was not driven by high tumor mutational burden (TMB). Instead, it is associated with presence of p53 mutations, absence of KRASG12 mutations, baseline genomic instability and proliferation gene signatures, as well as the presence of proliferating, tissue-resident CD8+ T cells with a dysfunctional and tumor-reactive phenotype. Transcriptomic analyses of pre- and post-treatment samples demonstrated consistent immune activation irrespective of clinical response. In contrast, a meta-analysis of over 450 patients across various tumor types treated mostly (90%) with neoadjuvant anti-CTLA-4 and anti-PD1 showed high expression of immune-related gene signatures correlated with response rates, as well as TMB and high proliferation signatures. Higher Wnt/β-catenin signatures were associated with non response. NEOASIS, a ‘basket’ pan-cancer study of anti-PD-1/anti-CTLA-4 for pMMR and dMMR tumors, including rare cancers and with an arm for any other cancers irrespective of known MMR status, is ongoing. In her concluding remarks, Chalabi underscored that MMR deficiency continues to be the most robust biomarker for predicting (neoadjuvant) immunotherapy success, advocating for its universal testing across tumor types to better pinpoint patients who may benefit. Furthermore, she stressed the critical role of compact, high-risk "window of opportunity" studies in yielding specialized data that can drive future clinical practice.
Reading suggestion:
Finding the right combination: new insights in anti-PD-1 plus anti-CTLA-4 or anti-LAG3
EXPANDing T Cells and their Tumor Antigens during Checkpoint Immunotherapy - Diether Lambrechts, KU Leuven, Belgium
Diether Lambrechts aimed to improve cancer immunotherapy by focusing on T cell dynamics in the tumor immune microenvironment (TIME), utilizing single cell TCR and RNA sequencing and spatial analysis, and ultimately using expanded TCR clonotypes in response to immune checkpoint blockade (ICB) to identity novel targets for vaccines. Two window-of-opportunity trials demonstrated clonotype expansion in response to ICB in some but not all patients, particularly among exhausted CD8+ T cells (CD8+Ex) and CD4+ Th1 (CD4+Th1) cells. Importantly, in the second trial (BELLINI), expansion was associated with clinical response, indicating the expanded clonotypes, and therefore antigens, were likely highly relevant. Examining the immune cell populations in multiple pan-cancer datasets and clustering the cell subtypes revealed the presence of tertiary lymphoid structures and type 1 immunity hubs, both of which correlated with T cell clonotype expansion in their studies in breast and head and neck cancers. Spatial transcriptome analysis confirmed the co-localization of these cell types, and both hubs could be observed in patients responding to ICB. Furthermore, CD8+Ex and CD4+Th1 cells were both proliferating in these hubs in samples treated with ICB. The key question Lambrechts posed was - What antigens are these cells responding to? - as understanding this could significantly improve cancer vaccine therapy, where ability to predict tumor control-relevant antigens, even neoantigens, is still problematic. To answer this, Lambrechts' team developed TWISTAR, an in vitro screening of T cells expressing TCR clonotypes against APCs expressing a representation of the full patient tumor transcriptome. Both the TCR-expressing line (GFP-positive) and the antigen-expressing APC line (RFP-positive) were highly optimized, which allowed dual color identification and isolation of both cell types by automated microscopy and cluster picking, followed by sequencing to identify TCR and antigen. As an example, using the cell line from a human melanoma brain metastasis and TCR clonotypes identified in the tumor, 50 TCR clonotypes were screened. 30 of the screened clonotypes were reactive to the cell line, and of these 30, 21 were reactive in the TWISTAR assay; all non-reactive clonotypes were negative by TWISTAR. All patient HLAs were involved, but of the 21 identified antigens, only 2 were due to somatic mutations and nearly half were from the 'dark' genome, representing non-canonical transcripts and translated regions, and the other nearly half were cancer-testis-antigens (e.g., MAGEA4) or differentiation antigens. Many of the unexpected translation products were derived from lncRNAs, many of which also showed a cancer-testis-like expression pattern. When TWISTAR was applied to CD8+ IFNγ+ expanding clonotypes from a breast cancer patient being treated with ICB, the majority of the TCRs recognized the 'dark' proteome, including one target that was shared across multiple cancer types. Similar results were observed in a ICB hyper-responsive patient with hepatocellular carcinoma, with another shared antigen detected. Following identification of the relevant epitopes using predictions and functional testing, the functional avidities of the detected antigens and expression levels spanned wide ranges, with neoantigen and lnc-derived antigens having the highest functional avidities. TWISTAR is being employed to select shared or personal antigens for vaccine trials.
By Ute Burkhardt, Ed Fritsch, and Lauren Hitchings



